Table of Contents
- Cytoscape 2.4.1 User Manual
- Introduction
- Launching Cytoscape
- Quick Tour of Cytoscape
- Command Line Arguments
- Cytoscape Preferences
- Creating Networks
- Supported Network File Formats
- Node and Edge Attributes
- Loading Gene Expression (Attribute Matrix) Data
- Navigation and Layout
- Visual Styles
- Finding and Filtering Nodes and Edges
- Editing Networks
- CytoPanels
- Rendering Engine
- Annotation
- Linkout
- Acknowledgements
- Appendix A: Old Annotation Server Format
- Appendix B: GNU Lesser General Public License
![]()
This document is licensed under the Creative Commons license, 2006
Authors: The Cytoscape Collaboration
The Cytoscape project is an ongoing collaboration between:
Cytoscape is a project dedicated to building open-source network visualization and analysis software. A software “Core” provides basic functionality to layout and query the network and to visually integrate the network with state data. The Core is extensible through a plug-in architecture, allowing rapid development of additional computational analyses and features.
Cytoscape's roots are in Systems Biology where it is used for integrating biomolecular interaction networks with high-throughput expression data and other molecular state information. Although applicable to any system of molecular components and interactions, Cytoscape is most powerful when used in conjunction with large databases of protein-protein, protein-DNA, and genetic interactions that are increasingly available for humans and model organisms. Cytoscape allows the visual integration of the network with expression profiles, phenotypes, and other molecular state information and to link the network to databases of functional annotations. The central organizing metaphor of Cytoscape is a network (graph), with genes, proteins, and molecules represented as nodes and interactions represented as links, i.e. edges, between nodes.
Development Cytoscape is a collaborative project between the Institute for Systems Biology (Leroy Hood lab), the University of California San Diego (Trey Ideker lab), Memorial Sloan-Kettering Cancer Center (Chris Sander lab), the Institut Pasteur (Benno Schwikowski lab), Agilent Technologies (Annette Adler lab) and the University of California, San Francisco (Bruce Conklin lab).
Visit http://www.cytoscape.org for more information.
License Cytoscape is protected under the GNU LGPL (Lesser General Public License). The License is included as an appendix to this manual, but can also be found online: http://www.gnu.org/copyleft/lesser.txt Cytoscape also includes a number of other open source libraries, which are detailed in Acknowledgements below.
What’s New in 2.4
Cytoscape version 2.4 contains several new features, plus improvements to the performance and usability of the software. These include:
- Publication quality image generation. This includes:
- Node label position adjustment.
- Automatic Visual Legend generator.
- Node position fine-tuning by arrow keys.
-
The ability to override selected VizMap settings.
-
Quick Find plugin.
- New Cytoscape icons for a cleaner user interface.
- Consolidated network import capabilities.
- Import network from remote data sources (through http or ftp).
- Default support for the following file formats:
- Delimited text table (TAB delimited text file, CSV, etc.)
-
Excel ( .xls ) format file.
-
New Ontology Server. This will eventually replace the BioDataServer. New Ontology Server features include:
- Native support for OBO format ontology files. Users can import many different ontologies in the OBO format.
- Ability to visualize the ontology tree as a network (DAG).
- Full support for Gene Association files.
- Support for Java SE 5.
- Many, many bug fixes!
Cytoscape is a Java application that runs on Linux, Windows, and Mac OS X.
System requirements: The system requirements for Cytoscape depend on the size of the networks the user wants to load, view and manipulate. We recommend a recent computer (1GHz CPU or higher) with a high-end graphics card and at least 512MB of free physical RAM. Cytoscape expects a minimum screen resolution of 1024x768.
If not already installed on your computer, download and install Java SE 5 or 6. Cytoscape 2.4 will no longer run with Java version 1.4.x or lower. You must install Java SE 5 or 6!!!
These can be found at:
There are a number of options for downloading and installing Cytoscape. All options can be downloaded from the http://cytoscape.org website.
- Automatic installation packages exist for Windows, Mac OS X, and Linux platforms.
- You can install cytoscape from a compressed archive distribution.
- You can build cytoscape from source.
-
You can check out the latest and greatest software from our Subversion repository.
Cytoscape installations (regardless of platform) containing the following files and directories:
Table 2.
|
File |
Description |
|
cytoscape.jar |
Main Cytoscape application (Java archive) |
|
cytoscape.sh |
Script to run Cytoscape from command line (Linux, Mac OS X) |
|
cytoscape.bat |
Script to run Cytoscape (Windows) |
|
LICENSE.txt/html |
Cytoscape GNU LGPL License |
|
lib/ |
library jar files needed to run Cytoscape. |
|
docs/ |
Manuals in different formats. What you are reading now. |
|
licenses/ |
Licence files for the various libraries distributed with Cytoscape. |
|
plugins/ |
Directory containing cytoscape PlugIns, in .jar format. |
|
sampleData/ |
|
|
galFiltered.gml -- Sample molecular interaction network file * |
|
|
galFiltered.sif -- Identical network in Simple Interaction Format * |
|
|
galExpData.pvals -- Sample gene expression matrix file * |
|
|
galFiltered.nodeAttrTable.xls -- Sample node attribute file in Microsoft Excel format |
|
|
galFiltered.cys -- Sample session file created from datasets above plus GO Annotations * |
|
|
BINDyeast.sif -- Network of all yeast protein-protein interactions in the BIND database as of Dec, 2006 ** |
|
|
BINDhuman.sif -- Network of all human protein-protein interactions in the BIND database as of Dec, 2006 ** |
|
|
yeastHighQuality.sif -- Sample molecular interaction network file *** |
|
|
interactome_merged.networkTable.gz -- Human interactome network file in tab-delimited format **** |
* From Ideker et al., Science 292:929 (2001)
** Obtained from data hosted at http://www.blueprint.org/bind/bind_downloads.html
** From von Mering et al., Nature, 417:399 (2002) and Lee et al, Science 298:799 (2002)
**** Created from Cytoscape tutorial web page. Original data sets are available at: http://www.cytoscape.orghttp://cytoscape.org/cgi-bin/moin.cgi/Data_Sets/ A merged human interactome by Andrew Garrow, Yeyejide Adeleye and Guy Warner Unilever, Safety and Environmental Assurance Center
Double-click on the icon created by the installer or by running cytoscape.sh from the command line (Linux or Mac OS X) or double-clicking cytoscape.bat (Windows). Alternatively, you can pass the .jar file to Java directly using the command java -Xmx512M -jar cytoscape.jar -p plugins. The -Xmx512M flag tells java to allocate more memory for Cytoscape and the -p plugins option tells cytoscape to load all of the plugins in the plugins directory. Loading the plugins is important because many key features like layouts, filters and the attribute browser are included with Cytoscape as plugins in the plugins directory. See the [Cytoscape_User_Manual/Command_Line_Arguments Command Line] chapter for more detail. In Windows, it is also possible to directly double-click the .jar file to launch it. However, this does not allow specification of command-line arguments (such as the location of the plugin directory).
Cytoscape Window When you succeed in launching Cytoscape, a window will appear that looks like this (captured on Linux):

For users interested in loading large networks, the amount of memory needed by Cytoscape will increase. Memory usage depends on both number of network objects (nodes+edges) and the number of attributes. Here are some rough suggestions for memory allocation:
Suggested Memory Size Without View
Table 3.
|
Number of Objects (nodes + edges) |
Suggested Memory Size |
|
0 - 70,000 |
512M (default) |
|
70,000 - 150,000 |
800M |
Suggested Memory Size With View
Table 4.
|
Number of Objects (nodes + edges) |
Suggested Memory Size |
|
0 - 20,000 |
512M (default) |
|
20,000 - 70,000 |
800M |
|
70,000 - 150,000 |
1G |
For the application to work properly, all files should be left in the directory in which they were unpacked. The core Cytoscape application assumes this directory structure when looking for the various libraries needed to run the application. If you are adventurous, you can get creative with the $CLASSPATH and/or the cytoscape.jar manifest file and run Cytoscape from any location you want.
When a network is loaded, Cytoscape will look something like the image below:

The main window here has several components:
- The menu bar at the top (See below for more information about each menu).
- The toolbar, which contains icons for commonly used functions. These functions are also available via the menus. Hover the mouse pointer over an icon and wait momentarily for a description to appear as a tooltip.
- The network management panel (top-left ). This contains an optional network overview pane (bottom-left overview of the network).
- The main network view window, which displays the network.
- The attribute browser panel (bottom panel), which displays attributes of selected nodes and edges and enables you to modify the values of attributes.
The network management and attribute browser panels are dockable tabbed panels known as CytoPanels. You can undock any of these panels by clicking on the Float Window control in the upper-right corner of the CytoPanel.
![]() If you select this control, e.g. on the attribute browser panel, you will now have two Cytoscape windows, the main window, and a new window labeled CytoPanel 2, similar to the one shown below.
If you select this control, e.g. on the attribute browser panel, you will now have two Cytoscape windows, the main window, and a new window labeled CytoPanel 2, similar to the one shown below.
 Note that CytoPanel 2 now has a Dock Window control. If you select this control, the window will dock onto the main window.
Note that CytoPanel 2 now has a Dock Window control. If you select this control, the window will dock onto the main window.
Cytoscape also has an editor that enables you to build and modify networks interactively by dragging and dropping nodes and edges from a palette onto the main network view window. The Node shapes and Edge arrows on the palette are defined by the currently used Visual Style. To edit a network, just select the Editor tab on CytoPanel 1. An example of an editor, with the palette contained in CytoPanel 1 and defined by the Bio Molecule Editor Visual Style, is shown below.

File The File menu contains most basic file functionality: File → Open for opening a Cytoscape session file, File → Save for saving a session file, File → Import for importing data such as networks and attributes, File → Export for exporting data and images. File → Print allows printing. File → Quit closes all windows of Cytoscape and exits the program. File → New creates a new network, either blank for editing, or from an existing network.

Edit The Edit menu contains Undo and Redo menu items which undo and redo edits made in the Attribute Browser and the Network Editor ONLY . Undo and Redo support do NOT work for other actions in Cytoscape such as layout.
There are also options for creating and destroying views (graphical representations of a network) and networks (the network data – not yet visualized), as well as an option for deleting selected nodes and edges from the current network. All deleted nodes and edges can be restored to the network via the Edit → Undo menu item. The Edit Menu also supports Preferences editing for properties and plug-ins via a Preferences Dialog.

View The View menu allows you to display or hide the network management panel (CytoPanel 1), the attribute browser (CytoPanel 2), the Network Overview (in CytoPanel 1), and the Advanced window. The View menu also allows you to open the VizMapper and lock the VizMapper. The View → Desktop provide further control over the various CytoPanels.

Select The Select menu contains different options selecting nodes and edges. It also contains the Select → Use Filters option which allows filters to be created which can be used to automatically select portions of a network whose node or edge attributes meet a filtering criterion.

Layout The Layout menu has an array of features for organizing the network visually. The top of the menu contains tools for manipulating sections of networks. These tools include scale, rotate, distribute, and align. The bottom section of the menu lists a variety of layout algorithms which automatically lay a network out.

Plugins The Plugins menu has menu items or choices added by plugins that have been loaded, such as "Import BioPAX Document from file". Depending on which plugins are loaded, the plugins that you see may be different than what appear here.

Table 5.
|
Note: A list of available Cytoscape Plugins with descriptions is available online at: http://cytoscape.org/plugins2.php |
Help The Help menu allows you to launch the online help viewer and browse the table of contents (Contents…). The “About…” menu item displays information about the running version of Cytoscape.

Cytoscape 2.3 allows multiple networks to be loaded at a time, either with or without a view. A network stores all the nodes and edges that are loaded by the user and a view displays them. You can have many views of the same network. Networks (and their optionally associated views) can be organized hierarchically.
An example where a number of networks have been loaded and arranged hierarchically is shown below:

The network manager (top-right tree view in CytoPanel 1) shows the networks that are loaded. Clicking on a network here will make that view active in the main window, if the view exists (green highlighted networks only). Each network has a name and size (number of nodes and edges), which are shown in the network manager. If a network is loaded from a file, the network name is the name of the file.
Some networks are very large (thousands of nodes and edges) and can take a long time to display. For this reason, a network in Cytoscape may not contain a ‘view’. Networks that have a view are highlighted in green and networks that don’t have a view are highlighted in red. You can create or destroy a view for a network by right-clicking the network name in the network manager or by choosing the appropriate option in the edit menu. You can also destroy previously loaded networks this way. In the picture above, seven networks are loaded, six green ones with views and one red one without a view.
Certain operations in Cytoscape will create new networks. If a new network is created from an old network, for example by selecting a set of nodes in one network and copying these nodes to a new network (via the File → New → Network option), it will be shown as a child of the network that it was derived from. In this way, the relationships between networks that are loaded in Cytoscape can be seen at a glance. Networks in the top part of the tree in the figure above were generated in this manner.
The available network views are also arranged as multiple, overlapping windows in the network view window. You can maximize, minimize, and destroy network views by using the normal window controls for your operating system.
The network overview window shows an overview (or ‘bird’s eye view’) of the network. It can be used to navigate around a large network view. This feature can be turned on or off via the View → Show/Hide Network Overview menu. The blue rectangle in the overview window shown below can be dragged with the mouse to navigate to a part of the network. The size of the navigation rectangle depends on the size of the active view and the zoom level of the view. The rectangle is smaller if the view is zoomed in and larger if zoomed out.

Table 6.
|
Important! The command line arguments have changed since version 2.2, so please read this section carefully. |
Cytoscape recognizes a number of optional command line arguments, including run-time specification of network files, attribute files, and session files. This is the output generated when the cytoscape is executed with the "-h" or "--help" flag.
usage: java -Xmx512M -jar cytoscape.jar [OPTIONS]
-h,--help Print this message.
-v,--version Print the version number.
-s,--session <file> Load a cytoscape session (.cys) file.
-N,--network <file> Load a network file (any format).
-e,--edge-attrs <file> Load an edge attributes file (edge attribute format).
-n,--node-attrs <file> Load a node attributes file (node attribute format).
-m,--matrix <file> Load a node attribute matrix file (table).
-p,--plugin <file> Load a plugin jar file, directory of jar files,
plugin class name, or plugin jar URL.
-P,--props <file> Load cytoscape properties file (Java properties
format) or individual property: -P name=value.
-V,--vizmap <file> Load vizmap properties file (Java properties format).
Any file specified for an option may be specified as either a path or as a URL. For example you can specify a network as a file (assuming that myNet.sif exists in the current working directory): cytoscape.sh -N myNet.sif. Or you can specify a network as a URL: cytoscape.sh -N http://example.com/myNet.sif.
Table 7.
|
Argument |
Description |
|
-h,--help |
This flag generates the help output you see above and exits. |
|
-v,--version |
This flag prints the version number of cytoscape and exits. |
|
-s,--session <file> |
This option specifies a session file to be loaded. Since only one session file can be loaded at a given time, this option may only specified once on a given command line. The option expects a .cys cytoscape session file. It is customary, although not necessary, for session file names to contain the .cys extension. |
|
-N,--network <file> |
This option is used to load all types of network files. SIF, GML, and XGMML files can all be loaded using the -N option. You can specify as many networks as desired on a single command line. |
|
-e,--edge-attrs <file> |
This option specifies an edge attributes file. You may specify as many edge attribute files as desired on a single command line. |
|
-n,--node-attrs <file> |
This option specifies a node attributes file. You may specify as many node attribute files as desired on a single command line. |
|
-m,--matrix <file> |
This option specifies a data matrix file. In a biological context, the data matrix consists of expression data. All data matrix files are read into node attributes. You may specify as many data matrix files as desired on a single command line. |
|
-p,--plugin <file> |
This option specifies a cytoscape plugin (.jar) file to be loaded by cytoscape. This option also subsumes the previous "resource plugin option". You may specify a class name that identifies your plugin and the plugin will be loaded if the plugin is in Cytoscape's CLASSPATH. For example, assuming that the class MyPlugin can be found in the CLASSPATH, you could specify the plugin like: |
|
-P,--props <file> |
This option specifies cytoscape properties. Properties can be specified either as a properties file (in Java's standard properties format), or as individual properties. To specify individual properties, you must specify the property name followed by the property value where the name and value are separated by the '=' sign. For example to specify the defaultSpeciesName: |
|
-V,--vizmap <file> |
This option specifies a visual properties file. |
Aside from plugins, all options described above can be loaded from the GUI once cytoscape is running.
Additional command line arguments that are not recognized by the Cytoscape core are passed to the Plugin modules. Please refer to the documentation for each specific Plugin for more details.
Table 8.
|
Important! If you have used previous versions of Cytoscape, you will notice that handling of properties has changed. The most important change is that properties are no longer saved by default to the current directory or to your home .cytoscape directory. Properties are stored by default in Cytoscape Session files. The cytoscape.props file still exists in the .cytoscape directory but is only written to when the user explicitly requests that the current settings be made the defaults for all future sessions of Cytoscape. Unless you have something important in your .cytoscape/cytoscape.props file, your best bet will be to delete the file and use the defaults. |
The Cytoscape Preferences Dialog, accessed via Edit → Preferences → Properties…, has sections for general properties display/editing and plugins specification via the properties mechanism. Preferences are now stored in Cytoscape session files. Any changes made to properties while running Cytoscape will be saved to the current session when you save the session. If you do not save the session, export the properties (File → Export), or set them as defaults (see below), the properties will be lost and the next time Cytoscape starts, defaults will be used.
Cytoscape properties are displayed in the Properties section of the dialog. These properties are configurable via Add, Modify and Delete operations.

Some common properties are described below.
Table 9.
|
Property name |
Default value |
Valid values |
|
defaultSpeciesName |
PleaseSpecify |
Species name. This value must match the name in the first line of the file specified in the bioDataServer’s manifest for synonyms e.g., for yeast synonyms, specify Saccharomyces cerevisiae |
|
bioDataServer |
PleaseSpecify |
annotation/manifest, and other manifest file locations |
|
viewThreshold |
10000 |
integer > 0 |
|
secondaryViewThreshold |
30000 |
integer > 0 |
|
viewType |
tabbed |
tabbed |
|
plugins |
comma-separated list of jar files containing plugins, or URL’s to jar files containing plugins (e.g., http://server/my-plugin.jar) |
|
|
defaultWebBrowser |
A path to the web browser on your system. This only needs to be specified if Cytoscape can’t find the web browser on your system. |
The specification of plugins to be loaded into Cytoscape at startup time is also supported in cytoscape.props and accessible in this dialog under the Plugins section. In this special case, the plugins property specifies a comma-separated list of jar files or URLs to jar files containing plugins. This property is parsed and presented and managed in the Plugins table, as at left.
Setting Default Properties It is possible to alter the default properties for Cytoscape. In the Cytoscape Preferences Dialog, accessed via Edit → Preferences → Properties…, edit any preferences, then click the "Make Current Cytoscape Properties Default" checkbox in the "Default Cytoscape Properties" section of the dialog. This will save any properties to the .cytoscape directory contained in your home directory. You should only do this if you want specific properties to apply to all of your Cytoscape sessions. You can rely on the Cytoscape session file to maintain the properties used for that particular session, so making certain properties default is not necessary to save the properties.
You can manage the available bookmarks on the system from the Edit → Preferences → Bookmarks… menu.
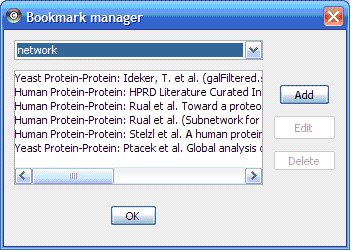
There are currently two types of bookmarks network and annotation . Network bookmarks are URLs pointing to network files available on the internet. These are nomal networks that can be loaded into Cytoscape. The annotation bookmarks are URLs pointing to ontology annotation files. The annotation bookmarks are only used when importing an ontology.
You can define and configure a proxy server for Cytoscape to use from the Edit → Preferences → Proxies… menu.

After the proxy server is set, all network traffic related to loading a network from URL will pass through the proxy server. Other plugins use this capability as well.
There are 3 different ways of creating networks in Cytoscape:
- Importing pre-existing, formatted network files.
- Importing pre-existing unformatted text or excel files.
- Creating an empty network and adding nodes and edges using the Editor.
Network files can be specified in any of the formats described in the [Cytoscape_User_Manual/Network_Formats Network Formats] chapter. Networks are imported into Cytoscape through the Import Network dialog, which can be activated through Menu File → Import → Network. The network file can either be located on the local computer, or it can also be located in a remote computer and referenced with a URL.
By default, Cytoscape load network from the local computer. When the import network dialog is initialized, the data source type “Local” will be selected. To import a network, first click on the “Select” button, which will pop-up a file chooser and let user browse the local disk to choose a network file. User is allowed only to choose Cytoscape recognized network types. After the file is selected, a click on the Import button will import the network. For example, The following steps will load a sample molecular interaction network in SIF format. Use the menu File → Import → Network. select the file “galFiltered.sif” in “sampleData” directory. After a few seconds, a small network of 331 nodes should appear in the main window. The procedure to load network in other format is very similar. To load the same interaction network as a GML, use the menu: File → Import → Network again. In the resulting file dialog box, select the file “sampleData/galFiltered.gml”. Node and edge attribute files as well as expression data and extra annotation can be loaded as well.
Network files may also be loaded directly from the command line using the –N option (works for SIF, GML, and XGMML).

Cytoscape also supports network import through URL. In this case, user should choose “Remote” as the data Source Type on the Import Network dialog. Next the user can either type (or paste) a URL into choice box or select one of the pre-existing bookmarked networks. Cytoscape supports URL bookmarks. A click on the right-most arrow of the comboBox will show a list of available network bookmarks. Cytoscape contains a pre-defined a list of bookmarks, which point to sample network files located on the Cytoscapes web server. Users may add new bookmarks through the Bookmark manager described in the [Cytoscape_User_Manual/Preferences Preferences] chapter.
After the user specifies a URL a click on the import button will load the network from the remote computer.

Network import from URL has an important caveat. Because Cytoscape determines file type primarily (not exclusively) by file extension, it can have trouble importing networks with URLs that don't end in a human readable file name. If Cytoscape does not recognize a meaningful file name with extension in the URL, it will attempt to guess the type of file based on MIME type. If the MIME type is not recognizable to any of our import handlers, then the import will fail.
Another issue for network import is the presence of firewalls, which can affect what files are accessible to a computer. To work around this problem, Cytoscape supports the use of proxy servers. To configure the proxy server, click Edit → Preference → Proxy Server…. This is further described in the [Cytoscape_User_Manual/Preferences Preferences] chapter.
Introduced in version 2.4, Cytoscape now supports the import of networks from delimited text files and excel files from the menu Edit → Import → Network from Table (Text/MS Excel).... An interactive GUI allows users to specify parsing options for specified files. The screen provides a preview that shows how the file will be parsed given the current configuration. As the configuration changes, the preview updates automatically. In addition to specifying how the file will be parsed, the user must also choose the columns that represent the Source nodes, the Target nodes, and an optional edge interaction type.
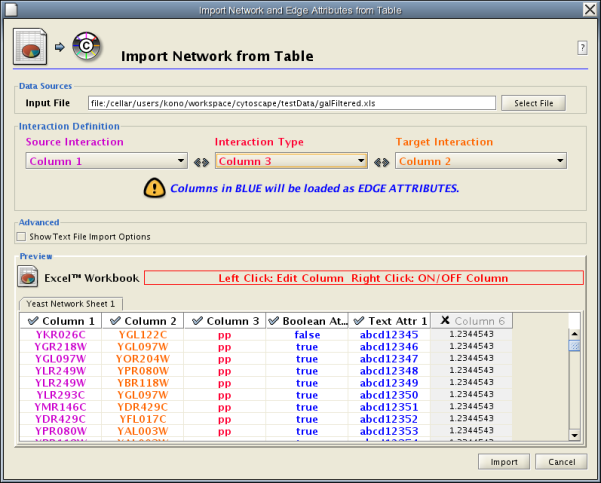
Network Table Import function supports delimited text files and Microsoft Excel Workbooks (1). The following is a sample table file:
source target interaction boolean attribute string attribute floating point attribute YJR022W YNR053C pp TRUE abcd12371 1.2344543 YER116C YDL013W pp TRUE abcd12372 1.2344543 YNL307C YAL038W pp FALSE abcd12373 1.2344543 YNL216W YCR012W pd TRUE abcd12374 1.2344543 YNL216W YGR254W pd TRUE abcd12375 1.2344543
The network table files should contain at least two columns: source nodes and target nodes. Interaction is optional in this format. Therefore, minimal network table looks like the following:
YJR022W YNR053C YER116C YDL013W YNL307C YAL038W YNL216W YCR012W YNL216W YGR254W
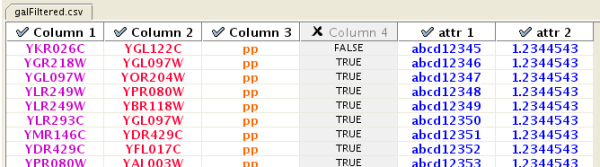
One row in a network table file represents an edge and its edge attributes. This means that a network file is considered a combination of network data and edge attributes. A table may contain columns that aren't meant to be edge attributes. In this case, you can choose not to import those columns by clicking on the column header in the preview window. This function is useful when importing a data table like the following (2):
Unique ID A Unique ID B Alternative ID A Alternative ID B Aliases A Aliases B Interaction detection methods First author surnames Pubmed IDs species A species B Interactor types Source database Interaction ID Interaction labels Cross-references Associated Files Experiment files Experiment labels Different techniques Different Pubmed articles Different sources Weight 7205 5747 TRIP6 PTK2 Q15654 Q05397-1 vv|HPRD Currently not available 14688263|15892868(Marcotte) Mammalia Homo sapiens protein|protein HPRD|Marcotte 0 Thyroid hormone receptor interactor 6-FAK-|PTK2-TRIP6 NA(HPRD)|NA(Marcotte) HPRD/02859_psimi.xml|other/ORIGINAL_DATA_MARCOTTE.txt vv(HPRD/02859_psimi.xml)|HPRD(other/ORIGINAL_DATA_MARCOTTE.txt) 17651(ExptRef)|Marcotte 2 2 2 2 4174 7311 MCM5 UBA52 P33992 P62987 neighbouring_reaction Currently not available 15608231(Reactome) Homo sapiens Homo sapiens protein|protein Reactome 1 P33992-P62988 Reaction:68944<->Reaction:68946(Reactome)|Reaction:68946<->Reaction:68944(Reactome) other/ORIGINAL_DATA_MARCOTTE.txt neighbouring_reaction(other/REACTOMEhomo_sapiens.interactions.txt) Reactome 1 1 1 1 7040 7040 TGFB1 TGFB1 P01137 P01137 nmr: nuclear magnetic resonance Currently not available 8679613 Homo sapiens Homo sapiens protein|protein BIND 2 TGFB1-TGFB1- 72085(BIND) BIND/bind_taxid9606.1.psi.xml nmr: nuclear magnetic resonance(BIND/bind_taxid9606.1.psi.xml) NotAvailable 1 1 1 1
This data file is a tab-delimited text and contains network data (interactions), edge attributes, and node attributes. To import network and edge attributes from this table, you need to choose Unique ID A as source, Unique ID B as target, and Interactor types as interaction type. Then you need to turn off columns used for node attributes ( Alternative ID A , species B , etc.). Other columns can be imported as edge attributes.
The network import dialog cannot import node attributes - only edge attributes. To import node attributes from this table, please see the Attributes section of this manual.
Note (1): in version 2.4, Cytoscape supports Excel Workbooks with single sheet (table) only. Multiple sheet Workbooks are not supported.
Note (2): from A merged human interactome datasets by Andrew Garrow, Yeyejide Adeleye and Guy Warner (Unilever, Safety and Environmental Assurance Center, 12 October 2006). Actual data files are available at:
http://www.cytoscape.orghttp://cytoscape.org/cgi-bin/moin.cgi/Data_Sets/
The last option for creating a network is to create a new, empty network and manual add nodes and edges. To do this, select the File → New → Network → Empty Network menu. This will create a new network. From here select the Edit tab in Cyto Panel 1 and edit the network as described in the [Cytoscape_User_Manual/Editor Cytoscape Editor] chapter.
Cytoscape can read network/pathway files written in the following formats:
- Simple interaction file (SIF or .sif format)
- Graph Markup Language (GML or .gml format)
- XGMML (extensible graph markup and modelling language).
- SBML
- BioPAX
- PSI-MI Level 1 and 2.5
- Delimited text
- Excel Workbook (.xls)
SIF specifies nodes and interactions only, while others store additional information about network layout and allows network data exchange with a variety of other network programs and data sources. Typically, SIF is used to import interactions when building a network for the first time, since it is easy to create in a text editor or spreadsheet. Once the interactions have been loaded and layout has been performed, the network may be saved to GML or XGMML format for interaction with other systems. All file types listed (except Excel) are text files and you can edit and view them in a regular text editor.
The simple interaction format is convenient for building a graph from a list of interactions. It also makes it easy to combine different interaction sets into a larger network, or add new interactions to an existing data set. The main disadvantage is that this format does not include any layout information, forcing Cytoscape to re-compute a new layout of the network each time it is loaded.
Lines in the SIF file specify a source node, a relationship type (or edge type), and one or more target nodes:
nodeA <relationship type> nodeB nodeC <relationship type> nodeA nodeD <relationship type> nodeE nodeF nodeB nodeG ... nodeY <relationship type> nodeZ
A more specific example is:
node1 typeA node2 node2 typeB node3 node4 node5 node0
The first line identifies two nodes, called node1 and node2, and a single relationship between node1 and node2 of type typeA. The second line specifies three new nodes, node3, node4, and node5; here "node2" refers to the same node as in the first line. The second line also specifies three relationships, all of type typeB and with node2 as the source, with node3, node4, and node5 as the targets, respectively. This second form is simply shorthand for specifying multiple relationships of the same type with the same source node. The third line indicates how to specify a node that has no relationships with other nodes. This form is not needed for nodes that do have relationships, since the specification of the relationship implicitly identifies the nodes as well.
Duplicate entries are ignored. Multiple edges between the same nodes must have different edge types. For example, the following specifies two edges between the same pair of nodes, one of type xx and one of type yy:
node1 xx node2 node1 xx node2 node1 yy node2
Edges connecting a node to itself (self-edges) are also allowed:
node1 xx node1
Every node and edge in Cytoscape has an identifying name, most commonly used with the node and edge data attribute structures. Node names must be unique as identically named nodes will be treated as identical nodes. The name of each node will be the name in this file by default (unless another string is mapped to display on the node using the visual mapper). This is discussed in the section on [Cytoscape_User_Manual/Visual_Styles visual styles]. The name of each edge will be formed from the name of the source and target nodes plus the interaction type: for example, sourceName (edgeType) targetName.
The tag <interaction type> can be any string. Whole words or concatenated words may be used to define types of relationships e.g. geneFusion, cogInference, pullsDown, activates, degrades, inactivates, inhibits, phosphorylates, upRegulates, etc.
Some common interaction types used in the Systems Biology community are as follows:
pp .................. protein – protein interaction pd .................. protein -> DNA (e.g. transcription factor binding upstream of a regulating gene.)
Some less common interaction types used are:
pr .................. protein -> reaction rc .................. reaction -> compound cr .................. compound -> reaction gl .................. genetic lethal relationship pm .................. protein-metabolite interaction mp .................. metabolite-protein interaction
Delimiters Whitespace (space or tab) is used to delimit the names in the simple interaction file format. However, in some cases spaces are desired in a node name or edge type. The standard is that, if the file contains any tab characters, then tabs are used to delimit the fields and spaces are considered part of the name. If the file contains no tabs, then any spaces are delimiters that separate names (and names cannot contain spaces).
If your network unexpectedly contains no edges and node names that look like edge names, it probably means your file contains a stray tab that's fooling the parser. On the other hand, if your network has nodes whose names are half of a full name, then you probably meant to use tabs to separate node names with spaces.
Networks in simple interactions format are often stored in files with a ".sif" extension, and Cytoscape recognizes this extension when browsing a directory for files of this type.
In contrast to SIF, GML is a rich graph format language supported by many other network visualization packages. The GML file format specification is available at:
http://www.infosun.fmi.uni-passau.de/Graphlet/GML/
It is generally not necessary to modify the content of a GML file directly. Once a network is built in SIF format and then laid out, the layout is preserved by saving to and loading from GML. Visual attributes specified in a GML file will result in a new visual style named “ Filename .style” when that GML file is loaded.
XGMML is the XML evolution of GML and is based on the GML definition. In addition to network data, XGMML contains node/edge/network attributes. The XGMML file format specification is available at:
http://www.cs.rpi.edu/~puninj/XGMML/
XGMML is now preferred to GML because it offers the flexibility associated with all XML document types. If you're unsure about which to use, choose XGMML.
BioPAX is an OWL (Web Ontology Language) document designed to exchange biological pathways data. Complete set of documents for this format is available at:
http://www.biopax.org/index.html
The PSI-MI format is a data exchange format for protein-protein interactions. It is an XML document to describe PPI and associated data. PSI-MI XML format specification is available at:
http://psidev.sourceforge.net/mi/xml/doc/user/
Cytoscape has native support for Microsoft Excel files (.xls) and delimited text files. The table in those file can have network data and edge attributes. Users can specify columns for source node, target node, interaction type, and edge attributes from GUI. For more detail, please read Import Free-Format Tables section.
Interaction networks are useful as stand-alone models. However, they are most powerful for answering scientific questions when integrated with additional information. Cytoscape allows the user to add arbitrary node, edge and network information to Cytoscape as node/edge/network(1) attributes . Attributes could be, for example, annotation data on a gene or confidence values in a protein-protein interaction. These attributes can then be visualized in a user-defined way by setting up a mapping from data attributes to visual attributes (colors, shapes, etc.) See the section on visual styles for a discussion of this.
Node and edge attribute files are simply formatted: A node attribute file begins with the name of the attribute on the first line, and on each following line, has the name of the node, followed by an equals sign, followed by the value of that attribute. Numbers and text strings are the most common attribute types. All values for a given attribute must have the same type. For example:
FunctionalCategory YAL001C = metabolism YAR002W = apoptosis YBL007C = ribosome
An edge attribute file has much the same structure, except that the name of the edge is the source node name, followed by the interaction type in parentheses, followed by the target node name. Directionality counts, so switching the source and target will refer to a different (or perhaps non-existent) edge. The following is an example edge attributes file:
InteractionStrength YAL001C (pp) YBR043W = 0.82 YMR022W (pd) YDL112C = 0.441 YDL112C (pd) YMR022W = 0.9013
Cytoscape treats edge attributes as directional, so note that the second and third edge attribute values refer to two different edges (source and target are reversed, though the nodes involved are the same).
Each attribute is stored in a separate file. Node and edge attribute files use the same format. Node attribute file names often use the suffix ".noa", while edge attribute file names use the suffix ".eda". Cytoscape recognizes these suffixes when browsing for attribute files.
Node and edge attributes may be loaded at the command line using the –n and –e options or via the File → Import menu.
When expression data is loaded using an expression matrix, it is automatically copied into the Node Attributes data structure unless explicitly specified not to.
Node and edge attributes are attached to nodes and edges, NOT to networks. If two different networks have the same nodes, then those nodes will have the same attributes. Even if a network is loaded after attributes have been loaded, if the nodes or edges found in the new network already exist, then any existing attributes will be applied to those nodes.
Note (1): Network attributes are supported in Cytoscape, but network attribute file reader is not yet implemented in Cytoscape 2.4. If you need to import network attributes, please use attribute table import function or write network attributes directly in XGMML file.
Every attribute file has one header line that gives the name of the attribute, and optionally some additional meta-information about that attribute. The format is as follows:
attributeName (class=formal.class.of.value)
The first field is always the attribute name: it cannot contain spaces. If present, the class field defines the formal (package qualified) name of the class of the attribute values. For example, java.lang.String for Strings, java.lang.Double for floating point values, java.lang.Integer for integer values, etc. If the value is actually a list of values, the class should be the type of the objects in the list. If no class is specified in the header line, Cytoscape will attempt to guess the type from the first value. If the first value contains numbers in a floating point format, Cytoscape will assume java.lang.Double; if the first value contains only numbers with no decimal point, Cytoscape will assume java.lang.Integer; otherwise Cytoscape will assume java.lang.String. Note that the first value can lead Cytoscape astray: for example,
floatingPointAttribute firstName = 1 secondName = 2.5
In this case, the first value will make Cytoscape think the values should be integers, when in fact they should be floating point numbers. It's safest to explicitly specify the value type to prevent confusion. A better format would be:
floatingPointAttribute (class=Double) firstName = 1 secondName = 2.5
or
floatingPointAttribute firstName = 1.0 secondName = 2.5
Every line past the first line identifies the name of an object (node in a node attribute file and an edge in a edge attribute file) along with the String representation of the attribute value. The delimiter is always an equals sign; whitespace (spaces and/or tabs) before and after the equals sign is ignored. This means that your names and values can contain whitespace, but object names cannot contain an equals sign and no guarantees are made concerning leading or trailing whitespace. Object names must be the Node ID or Edge ID as seen in the left-most column of the attribute browser if the attribute is to map to anything. These names must be reproduced exactly, including case, or they will not match.
Edge names are all of the form:
sourceName (edgeType) targetName
Specifically, that is
Table 10.
|
sourceName space openParen edgeType closeParen space targetName |
Note that tabs are not allowed in edge names. Tabs can be used to separate the edge name from the "=" delimiter, but not within the edge name itself. Also note that this format is different from the specification of interactions in the SIF file format. To be explicit: a SIF entry for the previous interaction would look like
sourceName edgeType targetName
or
Table 11.
|
sourceName whiteSpace edgeType whiteSpace targetName |
To specify lists of values, use the following syntax:
listAttributeName (class=java.lang.String) firstObjectName = (firstValue::secondValue::thirdValue) secondObjectName = (onlyOneValue)
This defines an attribute which is a list of Strings. The first object has three strings, and thus three elements in its list, while the second object has a list with only one member. In the case of a list every attribute value should be specified as a list, and every member of the list should be of the same class. Again, the class will be inferred if it is not specified in the header line. Lists are not supported by the visual mapper, so can’t be mapped to visual attributes.
Sometimes it is desirable to for attributes to include linebreaks, for example node labels that extend over two lines. You can acomplish by inserting the characters into the attribute value. For example:
newlineAttr YJL157C = This is a long\nline for a label.
Introduced in version 2.4, Cytoscape now supports importing delimited text and MS Excel attribute data tables. Using this functionality, users can now easily import data that isn't formatted into Cytoscape node or edge attribute file formats (as described above).

Sample Attribute Table 1
Table 12.
|
Object Key |
Alias |
SGD ID |
|
AAC3 |
YBR085W|ANC3 |
S000000289 |
|
AAT2 |
YLR027C|ASP5 |
S000004017 |
|
BIK1 |
YCL029C|ARM5|PAC14 |
S000000534 |
Attribute table file should contain a primary key column and at least one attribute column. Number of attribute columns is unlimited. Alias column is optional. First row can be used as attribute names, but it is optional. You can specify each attribute name from Attribute Table Import user interface.
User interface of Attribute Table Import is similar to Network Table Import .
-
Select File → Import → Attribute from Table (text/MS Excel)
-
Select one of the attribute types from Attributes radio buttons. Cytoscape can import node, edge, and network attributes.
-
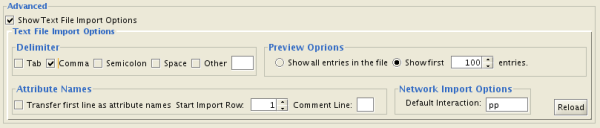
Select a data file. To load a local file, click on Select File button and choose a data file. Input file can be text or Excel (.xls) file. To load a remote file, type source URL directly in the text box. To show preview for the remote file, click Reload button on Advanced panel.
-
(Optional) If the table is not properly delimited, change delimiter from Text File Import Options panel. Default delimiter is TAB . This is not necessary for Excel Workbooks.
-
By default, the first column is set to primary key . Change the key column if necessary.
-
-
Click Import button.
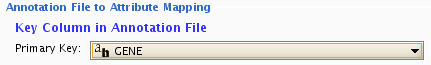
Attribute Table Import user interface has two advanced option panels to maximize mapping flexibility.
This panel is designed to change detail of mapping operation.
Primaly Key and Key Attribute
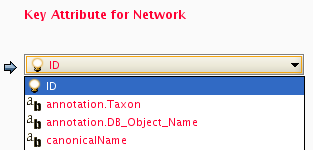
Old attribute file loader only supports mapping between node/edge ID and primary key in attribute file. To solve this limitation, new Attribute Table Import function supports both ID and attributes for mapping. You can choose an attribute for mapping from the list in the combo box.
Note: currently, only primitive data types (string, boolean, floating point, and integer) are supported for mapping, i.e. you cannot use list, map, or complex attribute as Key Attribute .
Aliases
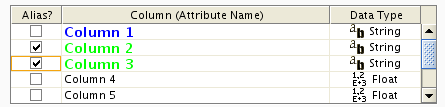
Cytoscape uses simple mechanism to manage aliases of objects. Both nodes and edges can have alias. If attributes are loaded as alias, they are treated as special attribute called alias . This will be used when mapping attributes. If primary key and key attribute for an object does not match, Cytoscape searches a match between aliases and key attribute. To use columns in attribute table as alias, just click on check boxes in the alias table.
-
This is mostly same as Network Table Import . For detail, please read Import Free-Format Table Files section in this manual.

When Cytoscape is started, the Attribute Browser appears in the bottom Cytopanel. This browser can be hidden and restored using the F5 key or the View → Show/Hide attribute browser menu option. Like other Cytopanels, the browser can be undocked by pressing the little icon in the browser’s top right corner.
To swap between displaying node, edge, and network attributes use the tabs on the bottom of the panel:
Node Attribute Browser
,
Edge Attribute Browser
, and
Network Attribute Browser
. The attribute browser displays attributes belonging to selected nodes and/or edges and the currently selected network. To populate the browser with rows (as pictured above), simply select nodes and/or edges in a loaded network. By default, only the
ID
of nodes and edges is shown. To display more than just the ID, click the
Select Attributes
![]() button and choose the attributes that are to be displayed. Each attribute chosen will result in one column in the attribute browser (in the screenshot above there are 5 columns total including the ID). Most attribute values can be edited by double-clicking an attribute cell; list values cannot be edited, and neither can the ID. Attribute rows in the browser can be sorted alphabetically by specific attribute by clicking on a column heading. A new attribute can be created using the
Create New Attribute
button and choose the attributes that are to be displayed. Each attribute chosen will result in one column in the attribute browser (in the screenshot above there are 5 columns total including the ID). Most attribute values can be edited by double-clicking an attribute cell; list values cannot be edited, and neither can the ID. Attribute rows in the browser can be sorted alphabetically by specific attribute by clicking on a column heading. A new attribute can be created using the
Create New Attribute
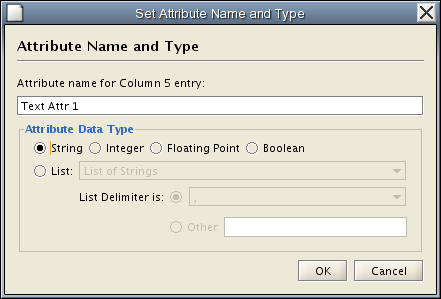
![]() button. Attributes created using the attribute browser must be one of four types – integer, string, real number (floating point), or boolean. Attributes can be deleted using the
Delete Attributes...
button. Attributes created using the attribute browser must be one of four types – integer, string, real number (floating point), or boolean. Attributes can be deleted using the
Delete Attributes...
![]() button. NOTE: Deleting attributes removes them from Cytoscape, not just the attribute browser! To remove attributes only from the browser simply unselect the attribute using the
Select Attributes
button. NOTE: Deleting attributes removes them from Cytoscape, not just the attribute browser! To remove attributes only from the browser simply unselect the attribute using the
Select Attributes
![]() button. The right-click menu on the Attribute Browser has several functions. This menu is useful for exporting attribute information to spreadsheet applications. For example, choose
Select All
and then
Copy
from the right-click and then paste into a spreadsheet application. Each attribute browser panel has a button for importing new attributes:
button. The right-click menu on the Attribute Browser has several functions. This menu is useful for exporting attribute information to spreadsheet applications. For example, choose
Select All
and then
Copy
from the right-click and then paste into a spreadsheet application. Each attribute browser panel has a button for importing new attributes:
![]() .
.
The Node Attribute Browser panel has additional buttons for loading Gene Expression attribute matrices (
![]() ) as node attributes.
) as node attributes.
In addition to normal node and edge attribute data, Cytoscape also supports importing gene expression data. Gene expression data are imported using a different file format than normal attributes, however the resulting attributes are no different from other attributes from Cytoscape's perspective. Gene expression data (like attribute data) can be loaded at any time, but are (generally) only relevant once a network has been loaded.
Gene expression ratios or values are specified over one or more experiments using a text file. Ratios result from a comparison of two expression measurements (experiment vs. control). Some expression platforms, such as Affymetrix, directly measure expression values, without a comparison. The file consists of a header and a number of space- or tab-delimited fields, one line per gene, with the following format:
Identifier [CommonName] value1 value2 ... valueN [pval1 pval2 ... pvalN]
Brackets [ ] indicate fields that are optional.
The first field identifies which Cytoscape node the data refers to. In the simplest case, this is the gene name - exactly as it appears on the Cytoscape canvas (case sensitive!). Alternatively, this can be some node attribute that identifies the node uniquely, such as a probeset identifier for commercial microarrays.
The next field is an optional common name. It is not used by Cytoscape, and is provided strictly for the user's convenience. With this common name field, the input format is the same as for commonly-used expression data anaysis packages such as SAM (http://www-stat.stanford.edu/~tibs/SAM/).
The next set of columns represent expression values, one per experiment. These can be either absolute expression values or fold change ratios. Each experiment is identified by its experiment name, given in the first line.
Optionally, significance measures such as P values may be provided. These values, generated by many microarray data analysis packages, indicate where the level of gene expression or the fold change appears to be greater than random chance. If you are using significance measures, then your expression file should contain them in a second set of columns after the expression values. The column names for the expression significance measures should match those of the expression values exactly .
For example, here is an excerpt from the file galExpData.pvals in the Cytoscape sampleData directory::
GENE COMMON gal1RG gal4RG gal80R gal1RG gal4RG gal80R YHR051W COX6 -0.034 0.111 -0.304 3.75720e-01 1.56240e-02 7.91340e-06 YHR124W NDT80 -0.090 0.007 -0.348 2.71460e-01 9.64330e-01 3.44760e-01 YKL181W PRS1 -0.167 -0.233 0.112 6.27120e-03 7.89400e-04 1.44060e-01 YGR072W UPF3 0.245 -0.471 0.787 4.10450e-04 7.51780e-04 1.37130e-05
This indicates that there is data for three experiments: gal1RG, gal4RG, and gal80R. These names appear two times: the first time gives the expression values, and the second gives the significance measures. For instance, the second line tells us that in Experiment gal1RG, the gene YHR051W has an expression value of -0.034 with significance measure 3.75720e-01.
Some variations on this basic format are recognized: see the formal file format specification below for more information. Expression data files commonly have the file extensions ".mrna" or ".pvals", and these file extensions are recognized by Cytoscape when browsing for data files.
COMMANDS: Load an expression attribute matrix file using the Cytoscape menu options File → Import → Attribute/Expression Matrix to bring up the import dialog box, or by specifying the filename using the -m option at the command line. If you use the command line input, you must enter your expression data by node ID. If you use the dialog box, then you can either load expression data by node ID (the default option), or can optionally select a node attribute to use in assigning your expression data to your Cytoscape nodes. If you do use a node attribute, then (1) the attribute should already be loaded, and (2) the node attribute value must match the first column in your matrix file.
For the sample network file sampleData/galFiltered.sif :
1. Load a sample gene expression data set using the menu: File → Import → Attribute/Expression Matrix. In the resulting file dialog box, in the field labeled "Please select an attribute or expression matrix file...", select sampleData/galExpData.pvals . The identifiers used in this file are the same ones used in the network file sampleData/galFiltered.sif , so you do not need to touch the field labeled "Assign values to nodes using...". A few lines of this file are shown below:
GENE COMMON gal1RG gal4RG gal80R gal1RG gal4RG gal80R YHR051W COX6 -0.034 0.111 -0.304 3.75720e-01 1.56240e-02 7.91340e-06 YHR124W NDT80 -0.090 0.007 -0.348 2.71460e-01 9.64330e-01 3.44760e-01 YKL181W PRS1 -0.167 -0.233 0.112 6.27120e-03 7.89400e-04 1.44060e-01
- or -
2a. After loading the network, load the node attribute file sampleData/gal.probeset.na , using the menu options File → Import → Node attributes.... This file is shown in part below:
Probeset YHR051W = probeset2 YHR124W = probeset3 YKL181W = probeset4
2b. After loading the node attribute file, select the expression data file sampleData.galExpPvals.probeset.pvals , shown in part below:
GENE COMMON gal1RG gal4RG gal80R gal1RG gal4RG gal80R probeset2 COX6 -0.034 0.111 -0.304 3.75720e-01 1.56240e-02 7.91340e-06 probeset3 NDT80 -0.090 0.007 -0.348 2.71460e-01 9.64330e-01 3.44760e-01 probeset4 PRS1 -0.167 -0.233 0.112 6.27120e-03 7.89400e-04 1.44060e-01
After selecting this file, in the field labeled Assign values to nodes using... , select Probeset . You will see that this loads exactly the same expression data as in Case 1, but provides extra flexibility in case the node name cannot be used as an identifier.
Detailed file format (Advanced users) In all expression data files, any whitespace (spaces and/or tabs) is considered a delimiter between adjacent fields. Every line of text is either the header line or contains all the measurements for a particular gene. No name conversion is applied to expression data files.
The names given in the first column of the expression data file should match exactly the names used elsewhere (i.e. in SIF or GML files).
The first line is a header line with one of the following three formats:
<text> <text> cond1 cond2 ... cond1 cond2 ... [NumSigConds] <text> <text> cond1 cond2 ... <tab><tab>RATIOS<tab><tab>...LAMBDAS
The first format specifies that both expression ratios and significance values are included in the file. The first two text tokens contain names for each gene. The next token set specifies the names of the experimental conditions; these columns will contain ratio values. This list of condition names must then be duplicated exactly, each spelled the same way and in the same order. Optionally, a final column with the title NumSigConds may be present. If present, this column will contain integer values indicating the number of conditions in which each gene had a statistically significant change according to some threshold.
The second format is similar to the first except that the duplicate column names are omitted, and there is no NumSigConds fields. This format specifies data with ratios but no significance values.
The third format specifies an MTX header, which is a commonly used format. Two tab characters precede the RATIOS token. This token is followed by a number of tabs equal to the number of conditions, followed by the LAMBDAS token. This format specifies both ratios and significance values.
Each line after the first is a data line with the following format:
FormalGeneName CommonGeneName ratio1 ratio2 ... [lambda1 lambda2 ...] [numSigConds]
The first two tokens are gene names. The names in the first column are the keys used for node name lookup; these names should be the same as the names used elsewhere in Cytoscape (i.e. in the SIF, GML, or XGMML files). Traditionally in the gene expression microarray community, who defined these file formats, the first token is expected to be the formal name of the gene (in systems where there is a formal naming scheme for genes), while the second is expected to be a synonym for the gene commonly used by biologists, although Cytoscape does not make use of the common name column. The next columns contain floating point values for the ratios, followed by columns with the significance values if specified by the header line. The final column, if specified by the header line, should contain an integer giving the number of significant conditions for that gene. Missing values are not allowed and will confuse the parser. For example, using two consecutive tabs to indicate a missing value will not work; the parser will regard both tabs as a single delimiter and be unable to parse the line correctly.
Optionally, the last line of the file may be a special footer line with the following format:
NumSigGenes int1 int2 ...
This line specified the number of genes that were significantly differentially expressed in each condition. The first text token must be spelled exactly as shown; the rest of the line should contain one integer value for each experimental condition.
Cytoscape uses a Zoomable User Interface for navigating and viewing networks. ZUIs use two mechanisms for navigation: zooming and panning. Zooming increases or decreases the magnification of a view based on how much or how little a user wants to see. Panning allows users to move the focus of a screen to different parts of a view.
Zoom Cytoscape provides two mechanisms for zooming, either using mouse gestures or buttons on the toolbar. Use the zooming buttons located on the toolbar to zoom in / out of the interaction network shown in the current network display. Zoom icons are detailed below:
From Left to Right:
- Zoom Out
- Zoom In
- Zoom Selected Region
- Zoom out to Display all of Current Network
You can also zoom in/out by holding down the right mouse button and moving the mouse to the right (zoom in) or left (zoom out).
Pan You can pan the network image by holding down the middle mouse button and moving the mouse. You can also pan the image by holding down the left mouse button over the blue box in the Network Overview panel in the lower left hand of the Cytoscape desktop.
The Layout menu has an array of features for organizing the network visually according to one of several algorithms, aligning and rotating groups of nodes, and adjusting the size of the network. Most of these features are available from plugins that are packaged with Cytoscape 2.3. Some of the layout algorithms provided with Cytoscape 2.3 are:
Cytoscape Spring-embedded Layout
Spring-embedded layout is based on a “force-directed” paradigm. Network nodes are treated like physical objects that repel each other, such as electrons. The connections between nodes are treated like metal springs attached to the pair of nodes. These springs repel or attract their end points according to a force function. The layout algorithm sets the positions of the nodes in a way that minimizes the sum of forces in the network. This algorithm is available from the Layout → Cytoscape Layouts → Spring Embedded menu item. A sample screen shot is provided below:
 yFiles Circular Layout
yFiles Circular Layout
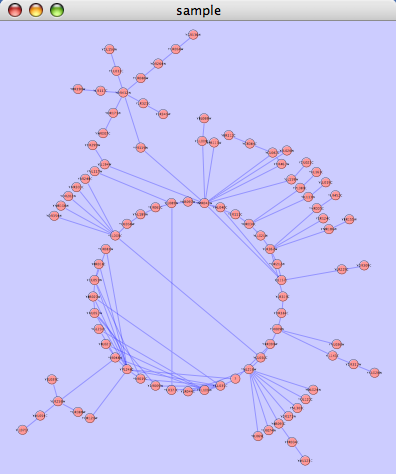
This algorithm produces layouts that emphasize group and tree structures within a network. It partitions the network by analyzing its connectivity structure, and arranges the partitions as separate circles. The circles themselves are arranged in a radial tree layout fashion. This algorithm is available from the Layout → yFiles → Circular menu item.
 yFiles Hierarchical Layout
yFiles Hierarchical Layout
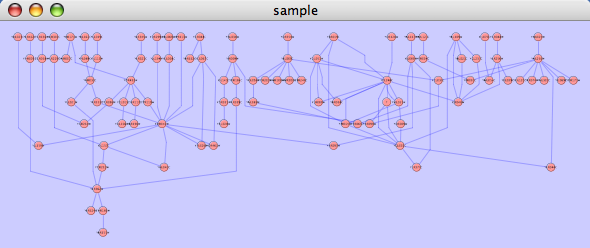
The hierarchical layout algorithm is good for representing main direction or flow within a network. Nodes are placed in hierarchically arranged layers and the ordering of the nodes within each layer is chosen in such a way that minimizes the number of edge crossings. This algorithm is available from the Layout → yFiles → Heirarchical menu item.
 yFiles Organic Layout
yFiles Organic Layout
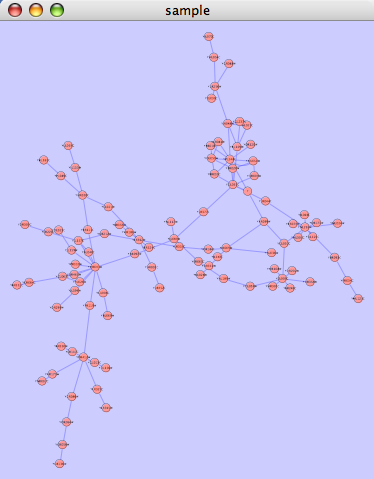
The organic layout algorithm is a kind of spring-embedded algorithm that combines elements of the other algorithms to show the clustered structure of a graph. This algorithm is available from the Layout → yFiles → Organic menu item.
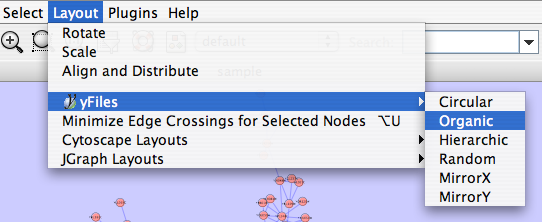
Several other alignment algorithms, including a selection from the JGraph project (http://jgraph.sourceforge.net), are also available under the Layout menu.
The simplest method to manually layout a network is to click on a node and drag the node. If you select multiple nodes, all of the selected nodes will be moved together.
Edge Handles
A little known feature! If you select an edge and then Ctrl-left-click on the edge, an edge "handle" will appear. This handle can be used to change the shape of the line. To remove a handle, simply Ctrl-left-click on the handle again.

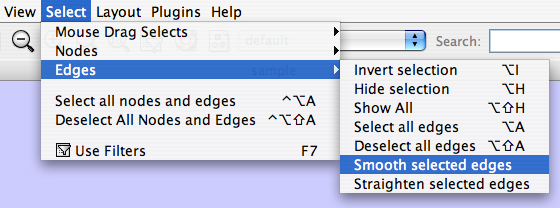
The Select → Edges menu has two menu items that provide further control: "Smooth Selected Edges" turns an edge consisting of line segments into a smoothed bezier curve, and "Straighen Selected Edges" turns a curved edge back into line segments.
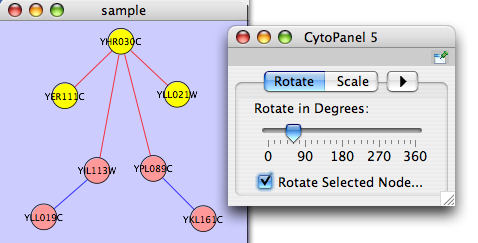
Rotate
The Layout → Rotate menu opens the Rotate dialog. Rotate will either rotate the entire network or a selected portion of the network. The image below shows a network with selected nodes rotated.
Before
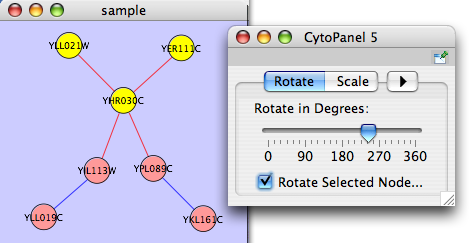
After
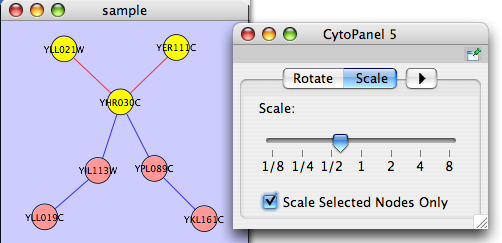
Scale
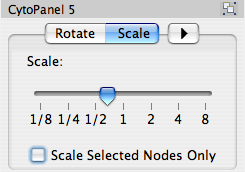
The Layout → Scale menu opens the Scale dialog. Scale will scale the position of the entire network or of the selected portion of the network. Note that only the position of the nodes scale, not the node sizes. Node size can be adjusted using the VizMapper. The image below shows selected nodes scaled.
Before
After
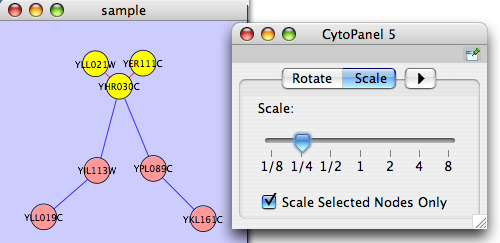
Align/Distribute
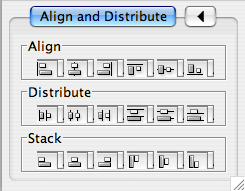
The Layout → Align/Distribute menu opens the Align/Distribute dialog. The Align buttons of the dialog provides different options for either vertically or horizontally aligning selected nodes against a line. The differences are in what part of the node gets aligned, e.g. the center of the node, the top of the node, the left side of the node. The Distribute buttons evenly distribute selected nodes between the two most distant nodes along either the vertical or horizontal axis. The differences are again a function what part of the node is used as a reference point for the distribution. The table below provides a mapping from button to name to result.
Table 13.
|
Button |
Before |
After |
Description of Align Options |
|
|
|
|
Vertical Align Top - The tops of the selected nodes are aligned with the top-most node. |
|
|
|
|
Vertical Align Center - The centers of selected nodes are aligned along a line defined by the midpoint between the top and bottom-most nodes. |
|
|
|
|
Vertical Align Bottom - The bottoms of the nodes are aligned with the bottom-most node. |
|
|
|
|
Horizontal Align Left - The left hand sides of the selected nodes are aligned with the left-most node. |
|
|
|
|
Horizontal Align Center - The centers of selected nodes are aligned along a line defined by the midpoint between the left and right-most nodes. |
|
|
|
|
Horizontal Align Right - The right hand sides of the selected nodes are aligned with the right-most node. |












Table 14.
|
Button |
Before |
After |
Description of Distribute Options |
|
|
|
|
Vertical Distribute Top - The tops of the selected nodes are distributed evenly between the top-most and bottom-most nodes, which should stay stationary. |
|
|
|
|
Vertical Distribute Center - The centers of the selected nodes are distributed evenly between the top-most and bottom-most nodes, which should stay stationary. |
|
|
|
|
Vertical Distribute Bottom - The bottoms of the selected nodes are distributed evenly between the top-most and bottom-most nodes, which should stay stationary. |
|
|
|
|
Horizontal Distribute Left - The left hand sides of the selected nodes are distributed evenly between the left-most and rigth-most nodes, which should stay stationary. |
|
|
|
|
Horizontal Distribute Center - The centers of the selected nodes are distributed evenly between the left-most and rigth-most nodes, which should stay stationary. |
|
|
|
|
Horizontal Distribute Right - The right hand sides of the selected nodes are distributed evenly between the left-most and rigth-most nodes, which should stay stationary. |












In addition to the ability to click on a node and drag it to a new position, Cytoscape now has the ability move nodes using the arrow keys on the keyboard. By selecting one or more nodes using the mouse and clicking one of the arrow keys (←, →, ↑, ↓) the selected nodes will move one pixel in the chosen direction. If an arrow key is pressed while holding the shift key down, the selected nodes will 10 pixels in the chosen direction.
Mouse movement has also been enhanced. If the shift key is held down while dragging a node, the node will only move horizontally, vertically, or along a 45 degree diagonal.
With the Cytoscape Visual Style feature, you can easily customize the visual appearance of your network. For example, you can:
- specify a default color and shape for all nodes.
- use specific line types to indicate different types of interactions, or
- visualize gene expression data along a color gradient.
All these features are available by selecting the View → Open Viz Mapper menu item or clicking on the Viz Mapper icon
 button on the main button bar.
button on the main button bar.
The Cytoscape distribution includes several predefined visual styles to get you started. To demonstrate these styles, try out the following example:
-
Load a sample network: From the main menu, select File → Import → Network, and select sampleData/galFiltered.sif.
-
Layout the network: select Layout → yFiles → Organic.
-
Load a sample set of expression data: From the main menu, select File → Import → Attribute Matrix and select sampleData/galExpData.pvals.
By default, the Visual Style labeled “default” will be automatically applied to your network. This default style has a blue background, circular pink nodes, and blue edges (see sample screenshot below).
 Figure: Using the default Visual Style.
Figure: Using the default Visual Style.
You can change visual styles by making a selection from the Visual Style pull down menu (available directly to the right of the
icon).
For example, if you select “Sample1”, a new visual style will be applied to your network, and you will see a white background and round blue nodes. Additionally, if you zoom in closer, you can see that protein-DNA interactions (specified with the label: pd) are drawn with dashed red edges, whereas protein-protein interactions (specified with the label: pp) are drawn with a light blue color (see sample screenshot below).
 Figure: Using the Sample1 Visual Style. Protein-Protein interactions (solid blue lines) are now distinguishable from Protein-DNA interactions (dashed red lines).
Figure: Using the Sample1 Visual Style. Protein-Protein interactions (solid blue lines) are now distinguishable from Protein-DNA interactions (dashed red lines).
Finally, if you select “Sample2”, gene expression values for each node will be colored along a color gradient between red and green (where red represents a low expression ratio, and green represents a high expression ratio - with thresholds set for the gal1RGexp experiment bundled with Cytoscape in the sampleData/galExpData.pvals file). See sample screenshot below:

Figure: Using the Sample2 Visual Style. Gene expression values are now displayed along a red/green color gradient.
The Cytoscape Visual Mapper has three core components: visual attributes, network attributes and visual mappers :
-
A visual attribute is any visual setting that can be applied to your network. For example, you can change all nodes to squares by setting the node shape visual attribute.
-
A network attribute is any attribute associated with a node or an edge. For example, each edge in a network may be associated with a label, such as “pd” (protein-DNA interactions), or “pp” (protein-protein interactions).
-
A visual mapper maps network attributes to visual attributes. For example, a visual mapper can map all protein-DNA interactions to the color blue, and all protein-protein interactions to the color red.
Cytoscape includes a large number of visual attributes. These are summarized in the tables below.
Table 15.
|
Visual Attributes Associated with Nodes: |
|
Node Color |
|
Node Border Color |
|
Node Border Line Type. The following options are available: |
|
|
|
Node Shape. The following options are available: |
|
|
|
Node Size: width and height of each node. |
|
Node Label: the text label for each node. |
|
Node Label Position: the posiiton of the label relative to the node. |
|
Node Font: node font and size. |


Table 16.
|
Visual Attributes Associated with Edges: |
|
Edge Color |
|
Edge Line Type. The following options are available: |
|
|
|
Edge Source Arrow. The following options are available: |
|
|
|
Edge Target Arrow. The following options are available: |
|
|
|
Edge Label: the text label for each edge. |
|
Edge Font: edge font and size. |

Table 17.
|
Global Visual Properties: |
|
Background Color |
For each visual attribute, you can specify a default value or define a visual mapping. Cytoscape currently supports three different types of visual mappers:
-
Passthrough Mapper: network attributes are passed directly through to visual attributes. A passthrough mapper only works for node / edge labels. For example, a passthrough mapper can draw the common gene name on all nodes.
-
Discrete Mapper: discrete network attributes are mapped to discrete visual attributes. For example, a discrete mapper can map all protein-protein interactions to the color blue.
-
Continuous Mapper : continuous graph attributes are mapped to visual attributes. Depending on the visual attribute, there are two types of continuous mappers:
-
continuous to continuous mapper : for example, you can map a continuous value (0..1) to a continuous color gradient (red..green) or node/font size (10..100).
-
continuous to discrete mapper : for example, all values below 0 are mapped to square nodes, and all values above 0 are mapped to circular nodes. However, there is no way to smoothly morph between circular nodes and square nodes.
-
The matrix below shows visual mapper support for each visual property.
Table 18.
|
Node Properties |
Passthrough Mapper |
Discrete Mapper |
Continuous Mapper |
|
Node Color |
- |
X |
X |
|
Node Border Color |
- |
X |
X |
|
Node Border Type |
- |
X |
o |
|
Node Shape |
- |
X |
o |
|
Node Size |
- |
X |
X |
|
Node Label |
X |
X |
o |
|
Node Font Family |
- |
X |
o |
|
Node Font Size |
- |
X |
X |
|
Edge Properties |
Passthrough Mapper |
Discrete Mapper |
Continuous Mapper |
|
Edge Color |
- |
X |
X |
|
Edge Line Type |
- |
X |
o |
|
Edge Source Arrow |
- |
X |
o |
|
Edge Target Arrow |
- |
X |
o |
|
Edge Label |
X |
X |
o |
|
Edge Font Family |
- |
X |
o |
|
Edge Font Size |
- |
X |
X |
Legend
Table 19.
|
Symbol |
Description |
|
- |
Mapping is not supported for specified visual property. |
|
X |
Mapping is fully supported for specified visual property. |
|
o |
Mapping is partially supported for specified visual property. Support for “continuous to continuous” mapping is not supported. |
To create a new visual style, select the View → Open Viz Mapper menu item, or select the VizMap icon in the main button bar. You will now see a new Visual Styles dialog box (shown below.)

Click the New button, and enter a name for your new visual style when prompted. Then click the Define button. You will now see the main Visual Styles Properties dialog box (shown below.)
From this dialog box, you can flip between Node Attributes, Edge Attributes, and Global Defaults. You can also specify default values for any visual property, or define a new custom mapping.
For example, to set the default node shape to triangles, select Node Attributes → Node Shape. Then, click the Change Default button, and select the Triangle icon from the selection list.
 To apply your visual style to your network, hit the
Apply to Network
button, available in the bottom of the dialog panel.
To apply your visual style to your network, hit the
Apply to Network
button, available in the bottom of the dialog panel.
The following tutorial demonstrates how to create a new visual style with a discrete mapper. The goal is to draw Protein-DNA interactions with blue edges, and Protein-Protein interactions with red edges.
-
Load a sample network: From the main menu, select File → Import → Network, and select sampleData/galFiltered.sif.
-
Select New to create a new Visual Style. Name your new style: “Sample3”.
-
Click the Define button to define the newly created Visual Style.
-
In the “Set Visual Properties” Dialog box, select Edge Attributes → Edge Color.
-
Click the New button in the mapping panel.
- You will be prompted to select a mapping type: passthrough mapper, discrete mapper or continuous mapper (for an overview of the differences between these mappers, please refer to the text above) Select “discrete mapper”, and enter a descriptive name. For example, enter: Interaction_Type_Color.
- From the “Map Attribute” pull-down menu, select “interaction.” You should now see two buttons, one for pd (Protein-DNA interactions), and one for pp (Protein-Protein interactions).
- Click the “pd” button and select a blue color.
- Click the “pp” button and select a red color.
- Click the “Apply to Network" button.
You network should now show “pd” interactions in blue, and “pp” interactions in red. Sample screenshot is below

The following tutorial demonstrates how to create a new continuous mapper. The goal is to superimpose gene expression data onto a network, and to display gene expression values along a color gradient.
-
Load a sample network: From the main menu, select File → Import → Network, and select sampleData/galFiltered.sif.
-
Load a sample set of expression data: From the main menu, select File → Import → Attribute Matrix and select sampleData/galExpData.pvals.
- Select “New” to create a new Visual Style. Name your new style: “Sample4”.
- Click the “Define” button to define the newly created Visual Style.
-
In the “Set Visual Properties” Dialog box, select Node Attributes → Node Color.
- Click the New button in the mapping panel.
- You will be prompted to select a mapping type: passthrough mapper, discrete mapper or continuous mapper (for an overview of the differences between these mappers, please refer to the section above) Select “continuous mapper”, and enter a descriptive name. For example, enter: Color_Gradient.
- From the “Map Attribute” pull-down menu, select “gal1RGexp.”
- Click the “Add Point” button twice to add two data points.
- Set the first point to “-1”, Below = Yellow, Equal = White.
- Set the second point to “2”, Equal = Red, Above = Black.
- Click the “Apply to Network" button.
This visual mapper will set all nodes with a gal1RGexp value less than –1 to Yellow, and all nodes with a gal1RGExp value greater than 2 to Black. Additionally, all values between –1 and 2 will be painted with a white/red color gradient. Sample screenshot is below.
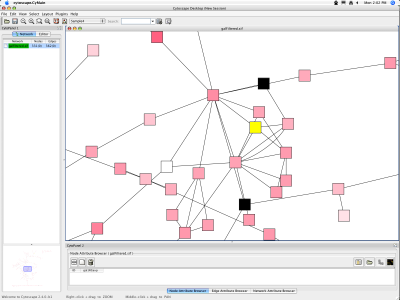
All Cytoscape Visual Style settings are initially loaded from a a default file called vizmap.props that cannot be altered by users. When users make changes to the visual properties, a vizmap.props file is saved in the session file. This means that assuming you save your session, you will not lose your visual properties. No other vizmap.props files are saved during normal operation.
Visual styles are automatically saved with the session they were created in. Before Cytoscape exits, you will be prompted to make sure you save the session before quitting. It is also possible to save your visual styles in a file separate from the session file. To do this, navigate to the File → Export → Vizmap Property File... menu and choose the file the properties should be saved to. This feature can be used to share visual styles with other users.
To import existing visual styles navigate to the File → Import → Vizmap Property File menu option and select a vizmap.props file. Imported properties will supplement existing properties or override existing properties if the properties have the same name. You can also specify a visual properties file using the -V command line option (cytoscape.sh -V myVizmap.props). Visual properties loaded from the command line will override any default properties.
It is possible to change the default visual properties for all sessions of cytoscape. To do this, navigate to the Edit → Preferences... menu, check the "Make Current Visual Styles Default" box in the "Default Visual Styles" section, and click "Ok". This will save the current visual styles as a vizmap.props file to your .cytoscape directory (found in your home directory). These visual styles will be loaded each time Cytoscape is started.
Cytoscape has a new feature that allows users to override visualizations created by the vizmapper for individual nodes and edges. This feature is available by right-clicking on a node or edge and then clicking on the Visual Mapping Bypass menu.

Each visual property of the node or edge is displayed. When a property is overridden, a checkmark appears next to the property and a [Reset <Property Name>] menu item appears below the checked property. By clicking the [Reset <Property Name>] option the bypass will be removed and the attribute will be displayed as defined by the VizMapper. At the bottom of the menu a Reset All option appears. When clicked, this will be remove all bypasses for the specified node or edge. In the example above you can see the the selected node size, color, and shape have been overridden. This is apparent in the appearance of the node itself and by the check marks in the popup menu.
It is important to realize that the the Visual Mapping Bypass only works for individual nodes and edges and not for all nodes or edges of a specific type. Using bypass is not particularly resource intensive, meaning you can use it as much as you like. However, if you ever find yourself repeating the same bypasses, then you should consider using the VizMapper instead.
Bypass is accomplished using special attributes with names like node.fillColor and node.shape . These are normal Cytoscape attributes and can be seen an editted in the Attribute Browser. The value of the attribute is a string representation of a property. For example, color is represented by 3 integers representing the RGB (red, green, blue) value of the color. Different types of properties have different string representations. When in doubt, just use the right click menu to create valid attribute values.
Because bypass values are specified using normal attributes, these attributes will persist between sessions as long as you save your session! If you don't save your session, you will lose whatever bypass values you set.
Cytoscape now includes a new Quick Find feature, which enables you to quickly find nodes and edges.
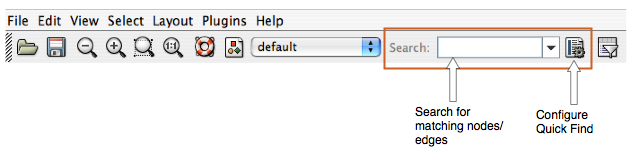
Using Quick Find is very simple. Here is how it works:
- Import up a network. For example, load up sampleData/galFiltered.sif
- Start typing in the text box. For example, enter "yd". The search box will automatically display a list of all matching nodes.
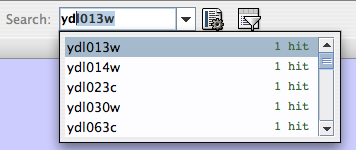
- Select a matching node by hitting [Enter]. Cytoscape will automatically zoom in on the selected node.
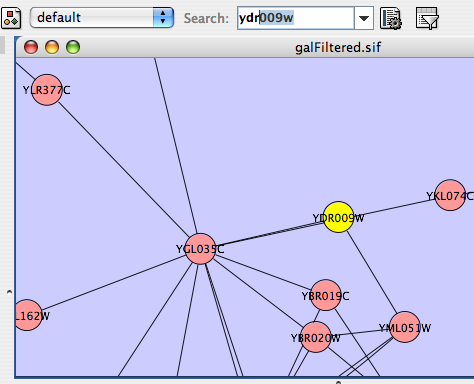
Quick Find works by creating an internal index of all nodes within the network. By default, Cytoscape indexes all nodes by the node identifier. However, you can configure Quick Find to index nodes or edges, and you can choose to index on any attribute.
For example, if you load up a BioPAX file (sampleData/Apoptosis_BioPAX.xml), your network will be automatically annotated with numerous attributes. To index the network based on, e.g. cellular location, click the Quick Find configuration button, and select "biopax_cellular" location from the drop-down menu.
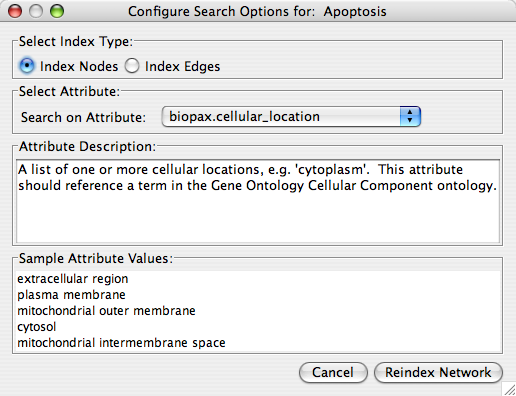
You can then quickly find all proteins located in the "plasma membrane" by just typing "p".
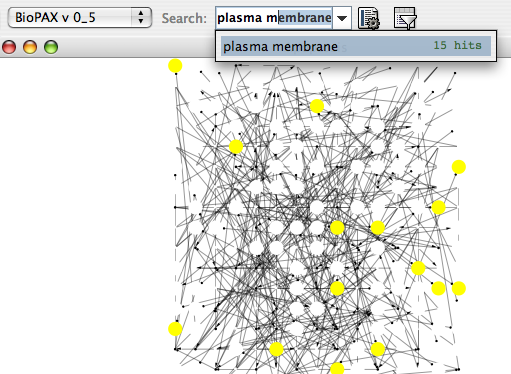
Tip: If you don't know what to search for, just leave the search box empty, and click on the down arrow directly next to the search box. Cytoscape will provide you with an initial list of matches. In the case below, we get a list of all distinct cellular locations in the network.
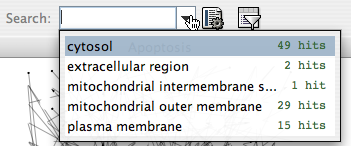
If you choose to index on a numerical attribute, the Cytoscape search box changes to a dynamic slider for quick filtering.
For example:
-
Import a network: sampleData/galFiltered.sif. File → Import → Network .
-
Import an expression data file: sampleData/galExpData.pvals. File → Import → Attribute / Expression Matrix
- Index Quick Find on the gal1RGExp attribute
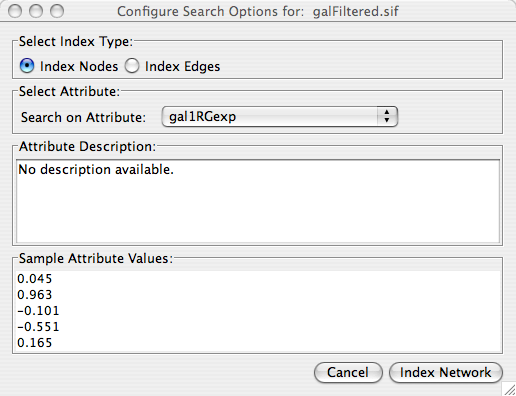
- Use the slider widget to automatically filter the entire network
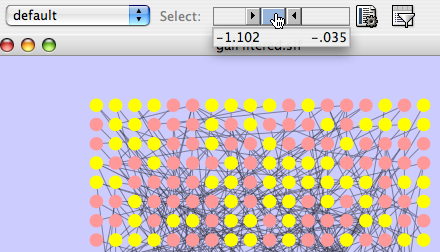
Filters allow for a wide variety of filtering on node and edge attributes loaded onto Cytoscape networks. Filters are poorly named because what they really do is select nodes or edges based on properties you specifiy. For example, you can select all the nodes whose name contains a specific pattern. Several types of filters are available. Basic filters allow the selection of multiple nodes or edges according to attribute data:
-
String filters allow selection of nodes or edges with attributes matching specified patterns. These patterns may include the wildcards * and ?.
-
Numerical filters allow selection of nodes or edges according to numerical attributes and the mathematical operators >, =, and <.
-
Topology filters allow selection of nodes with neighbors that match some pre-existing filter.
Compound filters allow selection based on the application of pre-existing filters:
-
Boolean filters allow the combination of multiple filters using the AND, OR and XOR operators. Example filters are shipped with the plugin to get started.
By default, you should see a filter icon on the toolbar:
![]()
If you press the filter icon, you will see a filters dialog which initially looks like the following:
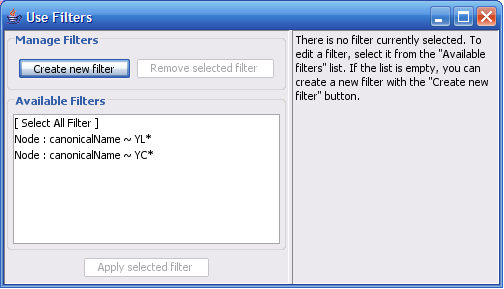
If the first filter is selected, then the dialog looks as shown:
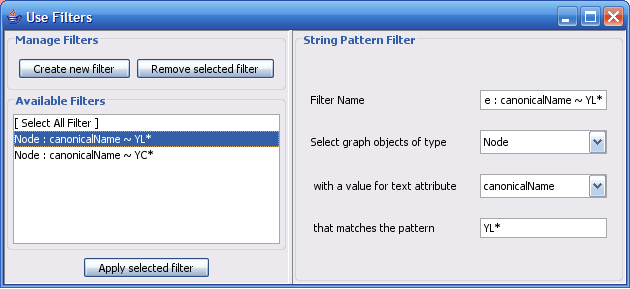
The right panel: An existing or newly created filter can be edited in this area. Each filter type has its own user interface for editing.
The low left panel: All available filters are shown in this list. Initially, this list will contain sample filters, but as you create more, they will be added here.
The up left panel: Pressing “Create new filter” adds a filter to the “Available Filters” box, and “Remove selected filter” deletes the currently selected filter.
The “Create new filter” button brings up the Filter creation dialog box, shown below.
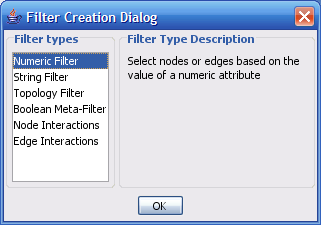
The important thing to realize when creating a filter is that the filter does not do anything by itself. Once created, the filter must be run.
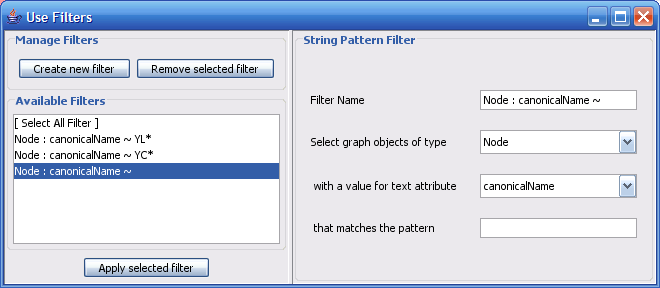
The String Filter allows you to filter nodes or edges by a given string node or edge attribute. Attributes that are loaded on the network are available for filtering against. Search terms are entered in the text box at the bottom. For example to match any Node whose canonicalName starts with “YDL” you would select “Node”, “canonicalName” and type “YDL*”. The * is important as it matches any number of characters after YDL. If you want to be more specific and only select nodes whose canonicalName starts with YDL00 followed by any other two characters, you would type “YDL00??”. The “?” denotes any single character, while the “*” represents zero or more characters. Full regular expression searching is supported, although is not covered here. Once the filter is defined, it will be assigned a default descriptive name.
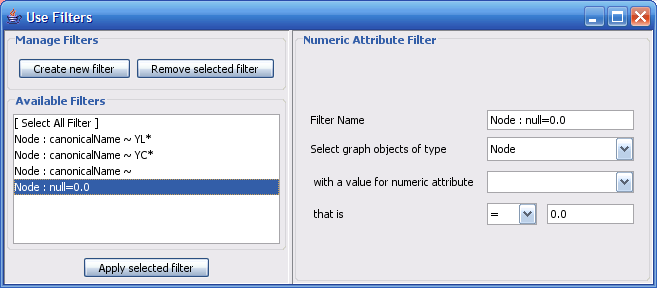
The Numerical Filter also allows you to filter nodes or edges, and presents you with a list of available attributes. This filter matches greater-than, less-than, or equal-to a number you type in the search box.
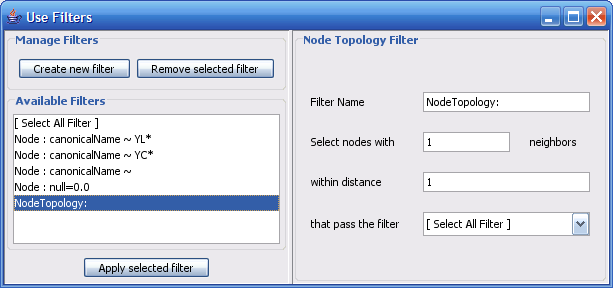
The node topology filter allows you to select nodes with at least n neighbors of distance m or less that pass some other selected filter. For instance, to select all the nodes adjacent to a node with the canonical name matching ‘YD*’, you would “select nodes with 1 neighbors”, “within distance 1”, “that pass the filter Node: canonicalName ~ YD*”.
The node topology filter depends on the existence of other filters. By default the [No Filter] is selected for this purpose. The [No Filter] doesn't filter anything, rather it selects all nodes.
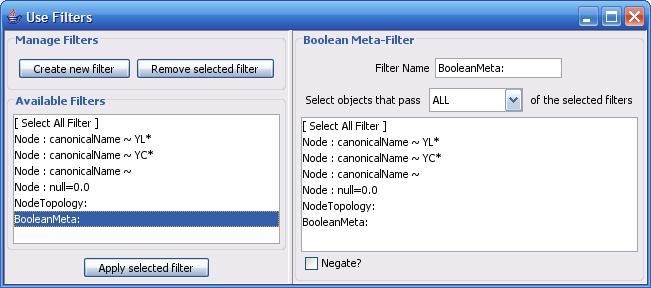
The Boolean Meta-Filter allows you to define a new filter that is a logical combination of existing filters. Available filters are displayed. By selecting one or more filters, you can then choose whether Nodes or Edges pass “ALL” (AND), “AT LEAST ONE” (OR), or “ONLY ONE” (XOR) of the selected filters. Once created Boolean filters can then themselves be combined using the Boolean filter to create arbitrarily complex logical combinations of filters. Note that unlike the String and Numerical Filters, Boolean Filters will need to be assigned a name manually.
Filters are currently saved automatically in the filters.props file found in the .cytoscape directory, found in each user's home directory. Once created, filters are saved for future sessions, as long as you exit Cytoscape normally via the exit command in the File menu (i.e. not via ctrl-c on Linux).
Any available filter can be run by pressing the ‘Apply selected filter’ button. When a filter is applied and multiple nodes or edges are selected, all of the normal selection-related operations may be performed, such as Delete Selected Node/Edges, Copy To New Network, and Invert Selection.
The Select → Nodes and Select → Edges menus provide several mechanisms for selecting nodes and edges. Most options are fairly straightforward, however some need extra explanation.
Select → Nodes → By Name... selects nodes by the node identifier (ID). This is the value seen in the left-most column of the attribute browser. This does not change if the node label changes!
Select → Nodes → From File... selects nodes based on node identifiers found in a specified file. The file format is simply one node id per line:
Node1 Node2 Node3 ...
Using Cytoscape's Editor, you can build and modify networks interactively by dragging and dropping nodes and edges from a palette onto the main network view window. The palette contains a set of shapes (for nodes) and arrows (for edges). The shapes on the palette are defined by the current Visual Style, with Node Shape and Node Color mapping into the shape and color of a node, and Edge Target Arrow mapping into the target arrow of an edge. An example of an editor, with the palette contained in CytoPanel 1, is shown below.

To edit an existing network, just select the Editor tab in CytoPanel 1. To start editing a new network, use the File → New → Network → Empty Network menu item.
The figure below shows the editor with palette defined by "Default" visual style.

To add a node to a network, drag and drop a node shape from the palette onto the canvas.
To connect two nodes with an edge, drag and drop an arrow shape onto a node on the canvas. This node becomes the source node of the edge. Move the cursor and a rubber-banded line follows the cursor. As the cursor passes over another node, that node is highlighted and the rubber-banded line will snap to a connection point on that second node. Click the mouse while over this node and the connection is established.
You can abort the drawing of the edge by clicking on an empty spot on the palette.
Note that if you change the Visual Style, the palette used by the current network view will also change to be consistent with the mappings in the new Visual Style.
There is also an Edit → Connect Selected Nodes menu item that, when chosen, creates a clique amongst the selected nodes.
The editor provides accelerators for adding nodes and edges. Control-clicking at a position on the canvas creates a node at that position. The NODE_TYPE attribute of the node will be the same as the NODE_TYPE of the node most recently added, defaulting to "DefaultNode" type. In this manner, you can use control-clicking as a kind of "stamp" to add multiple nodes of the same type to the network. Control-clicking on a node on the canvas starts an edge with source at that node. Move the cursor and a rubber-banded line follows the cursor. As the cursor passes over another node, that node is highlighted and the rubber-banded line will snap to a connection point on that second node. Control-click the mouse again and the connection is established. The EDGE_TYPE attribute of the edge will be the same as the EDGE_TYPE of the edge most recently added, defaulting to "DefaultEdge" type. You can abort the drawing of the edge by control-clicking on an empty spot on the palette.
You can delete nodes and edges by selecting a number of nodes and edges, then selecting the Edit → Delete Selected Nodes and Edges menu item. You can recover any nodes and edges deleted from a network by selecting the Edit → Undo menu item. Note that this will restore all nodes and edges that were previously deleted from the network, not just those deleted by the most recent delete operation.



In Cytoscape 2.3 a new network rendering engine is being introduced. The goal of the rendering engine is to be able to display large networks (>10,000 nodes) yet retain interactive speed. To accomplish this goal a technique involving level of detail is being used. Based on the count of objects (nodes and edges) being rendered, an appropriate level of detail is chosen. For example, by default, node labels (if present) are only rendered when less than 100 nodes are visible because drawing text is a relatively expensive operation. This can create some unusual behavior. If the screen currently contains 98 nodes, node labels will be displayed. If you pan the screen, such that now 101 nodes are displayed, the node labels will disappear. As another example, if the sum of rendered edges and rendered nodes is greater than or equal to a default value of 2000, a very coarse level of detail is chosen, where edges are always straight lines, nodes are always rectangles, and no antialiasing is done. The default values used to determine these thresholds can be changed by setting properties in the Edit → Preferences menu. Beware! The greater these thresholds become, the slower performance will become. If you work with small networks (a few hundred nodes), this shouldn't be a problem, but for large networks it will produce noticeable slowing. The various thresholds are described below.
Table 20.
|
render.coarseDetailThreshold |
If the sum of rendered nodes and rendered edges equals to or exceeds this number, a very coarse level of detail will be chosen and all other detail parameters will be ignored; this value defaults to 2000. |
|
|
render.nodeBorderThreshold |
If the number of rendered nodes equals to or exceeds this number, node borders will not be rendered; this value defaults to 200. |
|
|
render.nodeLabelThreshold |
If the number of rendered nodes equals to or exceeds this number, node labels will not be rendered; this value defaults to 100. |
|
|
render.edgeArrowThreshold |
If the number of rendered edges equals to or exceeds this number, edge arrows will not be rendered; this value defaults to 300. |
|
|
render.edgeLabelThreshold |
If the number of rendered edges equals to or exceeds this number, edge labels will not be rendered; this value defaults to 150. |
|
When printing networks or exporting to formats such as PostScript, the highest level of detail is always chosen regardless of what is currently being displayed on the screen.
Annotations in Cytoscape are stored as a set of ontologies (e.g. the Gene Ontology - GO). An ontology consists of a set of controlled vocabulary terms that annotate the objects. For example, using the Gene Ontology, the Saccharomyces Cerevisiae CDC55 gene’s biological process is described as “protein biosynthesis”, to which GO has assigned the number 6412 (a GO ID).
GO 8150 biological_process
GO 7582 physiological processes
GO 8152 metabolism
GO 44238 primary metabolism
GO 19538 protein metabolism
GO 6412 protein biosynthesis
Graphical View of GO Term 6412: protein biosynthesis
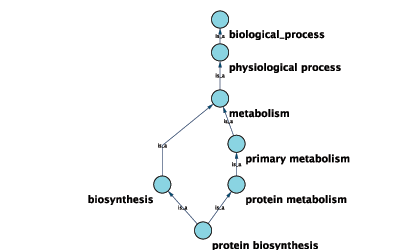
Cytoscape can use this ontology DAG (Directed Acyclic Graph) to annotate objects in networks. The Ontology Server (originally called Bio Data Server ) is a Cytoscape feature which allows you to load, navigate, and assign annotation terms to nodes and edges in a network. In version 2.4, Cytoscape has enhanced GUI for loading ontology and associated annotation, which enables you to load both local and remote files.
The standard file formats used in Cytoscape Ontology Server are OBO and Gene Association. The GO website details these file formats:
OBO file is the ontology DAG itself. This file defines relationships of ontology terms. Since version 2.4, Cytoscape can load all ontology files written in OBO format. The full listing of ontology files are available from Open Biomedical Ontologies (OBO) website:
Sample OBO File - gene_ontology.obo: http://www.geneontology.org/ontology/gene_ontology_edit.obo
format-version: 1.2 date: 27:11:2006 17:12 saved-by: midori auto-generated-by: OBO-Edit 1.002 subsetdef: goslim_generic "Generic GO slim" subsetdef: goslim_goa "GOA and proteome slim" subsetdef: goslim_plant "Plant GO slim" subsetdef: goslim_yeast "Yeast GO slim" subsetdef: gosubset_prok "Prokaryotic GO subset" default-namespace: gene_ontology remark: cvs version: $Revision: 5.49 $ [Term] id: GO:0000001 name: mitochondrion inheritance namespace: biological_process def: "The distribution of mitochondria, including the mitochondrial genome, into daughter cells after mitosis or meiosis, mediated by interactions between mitochondria and the cytoskeleton." [GOC:mcc, PMID:10873824, PMID:11389764] synonym: "mitochondrial inheritance" EXACT [] is_a: GO:0048308 ! organelle inheritance is_a: GO:0048311 ! mitochondrion distribution [Term] id: GO:0000002 name: mitochondrial genome maintenance namespace: biological_process def: "The maintenance of the structure and integrity of the mitochondrial genome." [GOC:ai] is_a: GO:0007005 ! mitochondrion organization and biogenesis
Default List of Ontologies
Cytoscape provides a list of ontologies available in OBO format. If Internet connection is available, Cytoscape imports ontology and annotation file directly from the remote source. The table below is the list of default ontoligies.
Table 21.
|
Ontology Name |
Description |
|
Gene Ontology Full |
This data source contains full size GO dag, which contains all GO terms. This OBO file is written in version 1.2 format. |
|
Generic GO slim |
Subset of general GO Terms. Includes higer-level terms only. |
|
Yeast GO slim |
Subset of GO Terms for annotating Yeast data sets. Maintained by SGD. |
|
Molecule role (INOH Protein name/family name ontology) |
A structured controlled vocabulary of concrete protein names and generic (abstract) protein names. This ontology is a INOH pathway annotation ontology, one of a set of ontologies intended to be used in pathway data annotation to ease data integration. This ontology is used to annotate protein names, protein family names, generic/concrete protein names in the INOH pathway data. INOH is part of the BioPAX working group. |
|
Event (INOH pathway ontology) |
A structured controlled vocabulary of pathway centric biological processes. This ontology is a INOH pathway annotation ontology, one of a set of ontologies intended to be used in pathway data annotation to ease data integration. This ontology is used to annotate biological processes, pathways, sub-pathways in the INOH pathway data. INOH is part of the BioPAX working group. |
|
Protein-protein interaction |
A structured controlled vocabulary for the annotation of experiments concerned with protein-protein interactions. |
|
Pathway Ontology |
The Pathway Ontology is a controlled vocabulary for pathways that provides standard terms for the annotation of geneproducts. |
|
PATO |
PATO is an ontology of phenotypic qualities, intended for use in a number of applications, primarily phenotype annotation. For more information, please visit PATO wiki (http://www.bioontology.org/wiki/index.php/PATO:Main_Page). |
|
Mouse pathology |
Mouse Pathology Ontology (MPATH) is an ontology for mutant mouse pathology. This is Version 1. |
|
Human disease |
This ontology is a comprehensive hierarchical controlled vocabulary for human disease representation. For more information, please visit Disease Ontology website (http://diseaseontology.sourceforge.net/). |
Although Cytoscape can import all kinds of ontologies in OBO format, you need to provide ontology-specific annotation file to annotate node/edge/network in Cytoscape . For example, while you can annotate human network data with GO Full and Gene Association file for human, you cannot annotate it with the combination of the Human disease ontology file and Gene Association file because the Gene Association file is annotation data only for GO.
-
Mapping Ontology - Annotation files are associated with a specific ontology. This means, you can map GO terms onto human network using Gene Association file for human, but cannot map Human Disease ontology terms with Gene Association file. In that case, you need an annotation file for Human disease ontology(see the diagram below):

Visualize and Browse Ontology DAG (for Advanced Users)
Relationships between ontology terms are usually represented as Directed Acyclic Graph (DAG). This is a special case of a network (or graph), where nodes are ontology terms and edges are relationships between terms. Originally, Cytoscape used special data structure called BioDataServer to store ontology DAGs. Since version 2.4, ontology data is stored in the same data structure as normal networks. This enables users and plugin writers to visualize, browse and manipulate ontology DAGs just like other networks. The following is an example of visualization of an ontology DAG (Generic GO Slim):
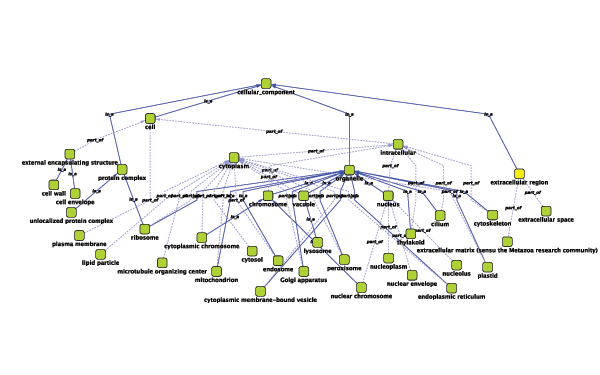
Every ontology term and relationship can have attributes. In the OBO files, ontology terms have optional fields such as definition, synonyms, comments, or cross-references. These fields will be imported as node attributes. To browse those attributes, please use attribute browser (see the example below):
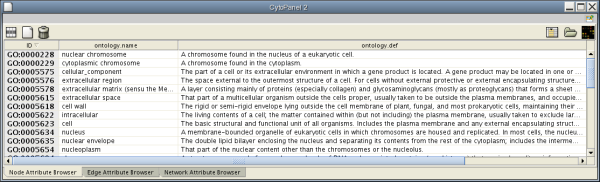
-
Note 1: Some ontologies have a lot of terms. For example, the full Gene Ontology contains more than 20,000 terms. If you need to save memory, you can remove this ontology DAG from Network Panel (Right click on the Ontology Name → Destroy Network ).
- Note 2: All ontology DAGs will be saved in the session file. To minimize the session file size, you can delete the Ontology DAG before saving session.
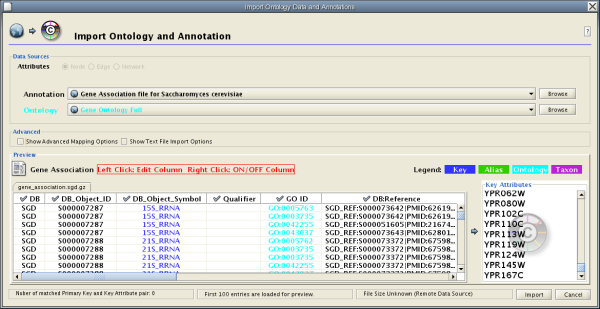 Cytoscape 2.4 provides a graphical user interface to import both ontology and annotation files at the same time.
Cytoscape 2.4 provides a graphical user interface to import both ontology and annotation files at the same time.
For user's convenience, Cytoscape has a list of URLs for commonly used ontology data and a complete set of Gene Association files. To import Gene Ontology and Gene Association files for the loaded networks, please follow these steps:
Important: All data sources in the preset list are remote URLs, meaning a network connection is required!
Step 1. Select Annotation File
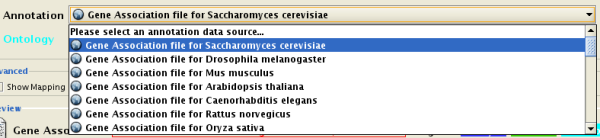
Select File → Import → Ontology and Annotation... to open the Import Ontology and Annotation dialog. From the Annotation combo box, select a gene association file for your network. For example, if you want to annotate yeast network, select Gene Association file for Saccharomyces cerevisiae .
Step 2. Select Ontology File
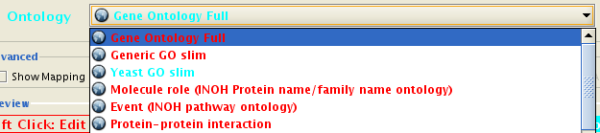
Select an Ontology data (OBO file) from Ontology combo box. If the file is not loaded yet, they are shown in red. The first three files are Gene Ontology files. You can load other ontologies, but you need your own annotation file to annotate networks.
Step 3. Start Import
Once you click Import button, Cytoscape start loading OBO and Gene Association files from the remote sources. If you choose GO Full it may take a while since it is a large data file.
Step 4.
When Cytoscape finishes to import files, the window will be automatically closed. All attributes mapped by this function have prefix annotation and looks like this: annotation .[attribute_name]. All ontologies will be added to the end of branch Ontology DAGs .

Ontology DAGs have some attributes associated with the terms. All attributes associated with ontology terms have prefix ontology. They have at least one attribute, ontology.name . For more informations about detail of attributes for ontology DAGs, please read OBO specification document.
- Note: Cytoscape supports both OBO format version 1.0 and 1.2.
Ontology and Annotation Import function is designed to import general ontology and annotation files. Internally, mapping ontology terms onto existing networks is same as joining three data tables in Cytoscape. An Ontology DAG, an annotation file, and networks are used in this process (see the example below).
Network Data

Ontology Data
Annotation Data
Mapping Result

If you want to map ontology terms onto network objects, you need to create a custom annotation file. The annotation file should contain at least 2 columns: a primary key and an ontology term ID . Primary key is the value for mapping between annotation file and network. Usually, node/edge ID is used as primary key, but you may choose any of the available attributes. Ontology term ID is the key for mapping between the ontology DAG and the annotation file. Using these data sources, you can annotate network objects in Cytoscape.
Suppose you have a small network:
node_1 pp node_2 node_3 pp node_1 node_2 pp node_3
and you want to annotate this network with Ontology A , which is an ontology DAG available in OBO format. In this case, you need an annotation table file looks like this:
node_1 OA_0000232 node_2 OA_0000441 node_3 OA_0000702
where OA_*** represents an ontology term ID. This example is a file with the minimum necessary number of columns, however you can include additional columns that will appear as additional node attributes.
Some ontologies will be used to annotate edges or networks. For example, Protein-protein interaction ontology is a controlled set of terms for annotating interactions between proteins, so ontology terms should be mapped onto edges (see example below).
node_1 (pp) node_2 MI:0445 node_3 (pp) node_1 MI:0046 node_2 (pp) node_3 MI:0346
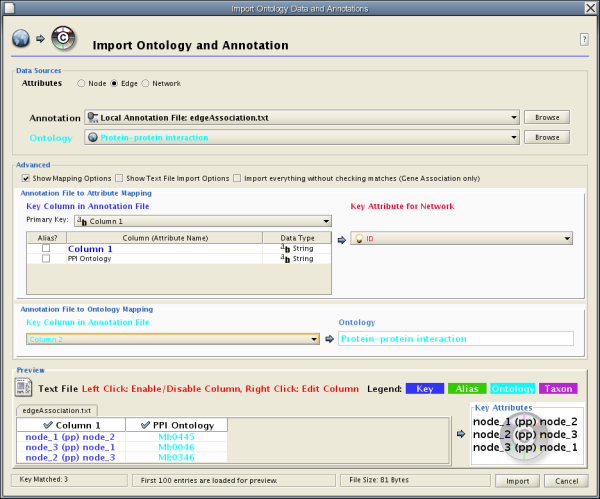
The basic operation of Ontology and Annotation Import is the same as Attribute Table Import . The main difference is that you need to specify an additional key for mapping:

By selecting a column from Key Column in Annotation File combo box, you can specify the key for mapping between ontology terms and annotation file.
-
Note: When you load Gene Association files, Cytoscape uses a special loader program designed only for Gene Association files. Because of this program, all attributes will be named automatically. Also, ontology IDs will be converted into term names and NCBI taxsonomy ID will be converted into actual species name. However, for custom annotation files, those processes will not be applied. All ontology terms will be mapped as term IDs.
LinkOut provides a mechanism to link nodes and edges to external web resources within Cytoscape. Right-clicking on a node or edge in Cytoscape view opens a popup menu with a list of web links.
The external links are specified in a linkout.props file which is included in the cytoscape.jar file. The defaults include a number of links such as Entrez, SGD, iHOP, Google, as well as a number of species-specific links. In addition to the default links, users can customize the LinkOut menu by adding (or removing) links by editing the linkout properties that can be found in the Edit → Preferences → Properties... dialog.
External links are listed as
‘key’-‘value’
pairs in the linkout.props file where
key
specifies the name of the link and
value
is the search URL. The LinkOut menus are organized in a hierarchical structure that is specified in the key. All key terms start with the keyword linkouturl.
For example, the following entry: linkouturl.yeast.SGD=http://db.yeastgenome.org/cgi-bin/locus.pl?locus\=%ID%
places the SGD link under the yeast submenu. This link will appear in Cytoscape as:

In a similar fashion one can added new submenus.
The
%ID%
string in the URL is a place-holder for the node label. When the popup menu is generated this marker is substituted with the node label. In the above example the generated SGD link for YIM protein is: http://db.yeastgenome.org/cgi-bin/locus.pl?locus\=YIM1. If you want to query based on a different attribute you currently need to specify a different Node Label using the VizMapper.
Currently there is no mechanism to check whether the constructed URL query is correct and if the node label is meaningful. Similarly there is no ID mapping between various identifiers. For example, a link to NCBI Entrez from a network that uses ensembl gene identifiers as node label will produce a link to Entrez using ensembl ID, which results in an incorrect link. It is the users responsibility to ensure that the node label that is used as the search term in the URL link will result in a meaningful link.
Adding or Removing links
The default links are defined in a linkout.props file contained in the linkout.jar. These links are normal java properties and can be editited while running in the Edit → Preferences → Properties... dialog. New links can be defined this way as well. New links can be defined at startup in a separate file and loaded from the command line, either by specifying a file containing the links cytoscape.sh -P new_linout.props or as individual properties cytoscape.sh -P linkouturl.yeast.SGD=http://db.yeastgenome.org/cgi-bin/locus.pl?locus\=%ID%. Any links defined on the command line will supersede the default links.
To remove a link from the menu simply delete the property from the Edit → Preferences → Properties... dialog.
Cytoscape is built with a number of open source 3rd party Java libraries. The Cytoscape team gratefully acknowledges the following libraries:
-
The Colt Distribution: Open Source Libraries for High Performance Scientific and Technical Computing in Java. Information is available at: http://hoschek.home.cern.ch/hoschek/colt/.
-
Graph INterface librarY a.k.a. GINY. Information is available at: http://csbi.sourceforge.net/.
-
JDOM. Information is available at: http://www.jdom.org.
-
JUnit. Information is available at: http://junit.org.
-
JGoodies Looks. Information is available at: http://www.jgoodies.com/freeware/looks/index.html.
-
Piccolo. Information is available at: http://www.cs.umd.edu/hcil/jazz/.
-
Type-Specific Collections Library, from Sosnoski Software Solutions, Inc. Information is available at: http://www.sosnoski.com/opensrc/tclib/.
-
Xerces Java XML parser. Information is available at: http://xml.apache.org/xerces-j/.
-
CLI command line parser. Information is available at: http://jakarta.apache.org/commons/cli/.
-
FreeHEP library. Information is available at: http://java.freehep.org.
This product includes software developed by the Apache Software Foundation (http://www.apache.org/).
This product includes software developed by the JDOM Project (http://www.jdom.org/).
One-step Installation of the Cytoscape software is accomplished using the InstallAnywhere product from ZeroG Software, Inc. (http://zerog.com)
Handler for the following format still exist in Cytoscape as a legacy code, however we strongly recommend using the new formats (OBO + Gene Association) described in the previous section, since they are easier to download directly from the Gene Ontology project and use directly. Currently, users have no access to import UI for this old format.
The annotation server requires that the gene annotations, and associated ontology on controlled vocabulary terms, follow a simple format. This simple format was chosen because it is efficient to parse and easy to use.
The flat file formats are explained below:
By example (the Gene Ontology - GO):
(curator=GO) (type=all) 0003673 = Gene_Ontology 0003674 = molecular_function [partof: 0003673 ] 0008435 = anticoagulant [isa: 0003674 ] 0016172 = antifreeze [isa: 0003674 ] 0016173 = ice nucleation inhibitor [isa: 0016172 ] 0016209 = antioxidant [isa: 0003674 ] 0045174 = glutathione dehydrogenase (ascorbate) [isa: 0009491 0015038 0016209 0016672 ] 0004362 = glutathione reductase (NADPH) [isa: 0015038 0015933 0016209 0016654 ] 0017019 = myosin phosphatase catalyst [partof: 0017018 ] ...
A second example (KEGG pathway ontology):
(curator=KEGG) (type=Metabolic Pathways) 90001 = Metabolism 80001 = Carbohydrate Metabolism [isa: 90001 ] 80003 = Lipid Metabolism [isa: 90001 ] 80002 = Energy Metabolism [isa: 90001 ] 80004 = Nucleotide Metabolism [isa: 90001 ] 80005 = Amino Acid Metabolism [isa: 90001 ] 80006 = Metabolism of Other Amino Acids [isa: 90001 ] 80007 = Metabolism of Complex Carbohydrates [isa: 90001 ] ...
The format has three required features:
-
The first line contains two parenthesized assignments, for curator and type . In the GO example above, the ontology file (which is created from the XML GO provides) nests all three specific ontologies (molecular function, biological process, cellular component) below the 'root' ontology, named 'Gene_Ontology'.
(type=all)
tells you that all three ontologies are included in that file.
- Following the mandatory title line, there are one or more category lines, each with the form:
number0 = name [isa:|partof: number1 number2 ...]
'isa' and 'partof' are terms used in GO; they describe the relation between parent and child terms in the ontology hierarchy.
-
The trailing blank before each left square bracket is not required; it is an artifact of the python script that creates these files.
By example (from the GO biological process annotation file):
(species=Saccharomyces cerevisiae) (type=Biological Process) (curator=GO) YMR056C = 0006854 YBR085W = 0006854 YJR155W = 0006081 ...
and from KEGG:
(species=Mycobacterium tuberculosis) (type=Metabolic Pathways) (curator=KEGG) RV0761C = 10 RV0761C = 71 RV0761C = 120 RV0761C = 350 RV0761C = 561 RV1862 = 10 ...
The format has these required features:
-
The first line contains three parenthesized assignments, for species , type and curator . In the example just above, the annotation file (which we create for budding yeast from the flat text file maintained by SGD for the Gene Ontology project, and available both at their web site and at GO's) shows three yeast ORFs annotated for biological process, with respect to GO, described (further above).
- Following the mandatory title line, there are one or more annotation lines, each with the form:
canonicalName = ontologyTermID
-
Once loaded, this annotation (along with the accompanying ontology) can be assigned to nodes in a Cytoscape network. For this to work, the species type of the node must exactly match the species named on the first line of the annotation file. The canonicalName of your node must exactly match the canonicalName present in the annotation file. If you don’t see the expected results when using this feature of Cytoscape, check this again, as getting either of these wrong is a common mistake.
The easiest way to make annotations available to Cytoscape is by loading annotations into the Cytoscape annotation server. This is the default behavior for the official release of Cytoscape.
You must first create a text file to specify the files you want Cytoscape to load. Here is an example, from a file which (for convenience) we usually call “manifest”
ontology=goOntology.txt annotation=yeastBiologicalProcess.txt annotation=yeastMolecularFunction.txt annotation=yeastCellularComponent.txt
Use the Cytoscape -b command line argument to specify the annotation manifest file to read (e.g. -b manifest). Please note that the -s switch, which sets the default species for your data is required to exactly match the species named in any annotation file you wish to use.
The Gene Ontology (GO) project is a valuable source of annotation for the genes of many organisms. In this section we will explain how to:
- Obtain the GO ontology file
- Reformat it into the simpler flat file Cytoscape uses
- Obtain an annotation file (we illustrate with yeast and human annotation)
- Reformat the annotation files into the simple Cytoscape format
Obtain the GO ontology file Go to the GO XML FTP (ftp://ftp.geneontology.org/pub/go/xml/) page. Download the latest go-YYYYMM-termdb.xml.gz file.
Reformat GO XML ontology file into a flat file
gunzip go-YYYYMM-termdb.xml.gz python parseGoTermsToFlatFile.py go-YYYYMM-termdb.xml > goOntology.txt
(See below for Python script listing)
Obtain the 'association' file for your organism GO maintains a list of association files for many organisms; these files associate genes with GO terms. The next step is to get the file for the organism/s you are interested in, and parse it into the form Cytoscape needs. A list of files may be seen at http://www.geneontology.org/GO.current.annotations.shtml. The rightmost column contains links to tab-delimited files of gene associations, by species. Choose the species you are interested in, and click 'Download'.
Let's use 'GO Annotations @ EBI Human' as an example. After you have downloaded and saved the file, look at the first few lines:
SPTR O00115 DRN2_HUMAN GO:0003677 PUBMED:9714827 TAS F Deoxyribonuclease II precursor IPI00010348 protein taxon:9606 SPTR SPTR O00115 DRN2_HUMAN GO:0004519 GOA:spkw IEA F Deoxyribonuclease II precursor IPI00010348 protein taxon:9606 20020425 SPTR SPTR O00115 DRN2_HUMAN GO:0004531 PUBMED:9714827 TAS F Deoxyribonuclease II precursor IPI00010348 protein taxon:9606 SPTR ...
Note that line wrapping has occurred here, so each line of the actual file is wrapped to two lines. The goal is to create from these lines the following lines:
(species=Homo sapiens) (type=Molecular Function) (curator=GO) IPI00010348 = 0003677 IPI00010348 = 0004519 IPI00010348 = 0004531 ...
or
(species=Homo sapiens) (type=Biological Process) (curator=GO) NP_001366 = 0006259 NP_001366 = 0006915 NP_005289 = 0007186 NP_647593 = 0006899 ...
The first sample contains molecular function annotation for proteins, and each protein is identified by its IPI number. IPI is the International Protein Index, which maintains cross references to the main databases for human, mouse and rat proteomes. The second sample contains biological process annotation, and each protein is identified by its NP (RefSeq) number. These two naming systems, IPI and RefSeq, are two of many that you can use for canonical names when you run Cytoscape. For budding yeast, it is much easier: the yeast community always uses standard ORF names, and so Cytoscape uses these as canonical names. For human proteins and genes, there is no such single simple standard. See section 5. Building and Storing Interaction Networks for more information.
The solution (for those working with human genes or proteins) is, once you have downloaded the annotations file, to:
- Decide which naming system you want to use.
-
Download ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/HUMAN/xrefs.goa. This cross-reference file, when used strategically, allows you to create Cytoscape-compatible annotation files in which the canonical name is the one most meaningful to you.
-
Examine xrefs.goa to figure out which column contains the names you wish to use.
- Make a very slight modification to the python script described below, and then
- Run that script, supplying both xrefs.goa and that annotation file as arguments.
Here are a few sample lines from xrefs.goa :
SP O00115 IPI00010348 ENSP00000222219; NP_001366; BAA28623;AAC77366;AAC35751;AAC39852;BAB55598;AAB51172;AAH10419; 2960,DNASE2 1777,DNASE2 SP O00116 IPI00010349 ENSP00000324567;ENSP00000264167; NP_003650; CAA70591; 327,AGPS 8540,AGPS SP O00124 IPI00010353 ENSP00000265616;ENSP00000322580; NP_005662; BAA18958;BAA18959;AAH20694; 7993,D8S2298E ...
Note that line wrapping has occurred here – each line in this example starts with the letters SP. See the README file for more information (ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/HUMAN/README)
Finally, run the script to create your three annotation files for human proteins:
-
bioproc.anno (GO biological process annotation)
-
molfunc.anno (GO molecular function annotation)
-
cellcomp.anno (GO cellular component annotation)
using the supplied python script. It may be necessary to modify this script slightly if RefSeq identifiers are not used as canonical names or if you are using a more recent version of Python.
python parseAssignmentsToFlatFileFromGoaProject.py gene_association.goa_human xrefs.goa
(See below for Python script listing)
Python script examples: These scripts, described above require Python version 2.2 or later.
Script 1 - parseGoTermsToFlatFile.py
# parseGoTermToFlatFile.py: translate a GO XML ontology file into a simpler
# Cytoscape flat file
#-----------------------------------------------------------------------------------
# RCS: $Revision: 1.3 $ $Date: 2003/05/18 00:38:43 $
#-----------------------------------------------------------------------------------
import re, pre, sys
#-----------------------------------------------------------------------------------
def flatFilePrint (id, name, isaIDs, partofIDs):
isa = ''
if (len (isaIDs) > 0):
isa = '[isa: '
for isaID in isaIDs:
isa += isaID
isa += ' '
isa += ']'
partof = ''
if (len (partofIDs) > 0):
partof = '[partof: '
for partofID in partofIDs:
partof += partofID
partof += ' '
partof += ']'
result = '~np~%~/np~s = ~np~%~/np~s ~np~%~/np~s ~np~%~/np~s' ~np~%~/np~ (id, name, isa, partof)
result = result.strip ()
if (result == 'isa = isa' or result == 'partof = partof'):
print >> sys.stderr, 'meaningless term: ~np~%~/np~s' ~np~%~/np~ result
else:
print result
#-----------------------------------------------------------------------------------
if (len (sys.argv) != 2):
print 'usage: ~np~%~/np~s <someFile.xml>' ~np~%~/np~ sys.argv [0]
sys.exit ();
inputFilename = sys.argv [1];
print >> sys.stderr, 'reading ~np~%~/np~s...' ~np~%~/np~ inputFilename
text = open (inputFilename).read ()
print >> sys.stderr, 'read ~np~%~/np~d characters' ~np~%~/np~ len (text)
regex = '<go:term .*?>(.*?)</go:term>';
cregex = pre.compile (regex, re.DOTALL) # . matches newlines
m = pre.findall (cregex, text)
print >> sys.stderr, 'number of go terms: ~np~%~/np~d' ~np~%~/np~ len (m)
regex2 = '<go:accession>GO:(.*?)</go:accession>.*?<go:name>(.*?)</go:name>'
cregex2 = re.compile (regex2, re.DOTALL)
regex3 = '<go:isa\s*rdf:resource="http://www.geneontology.org/go#GO:(.*?)"\s*/>'
cregex3 = re.compile (regex3, re.DOTALL)
regex4 = '<go:part-of\s*rdf:resource="http://www.geneontology.org/go#GO:(.*?)"\s*/>'
cregex4 = re.compile (regex4, re.DOTALL)
goodElements = 0
badElements = 0
print '(curator=GO) (type=all)'
for term in m:
m2 = re.search (cregex2, term)
if (m2):
goodElements += 1;
id = m2.group (1)
name = m2.group (2)
isaIDs = []
m3 = re.findall (cregex3, term);
for ref in m3:
isaIDs.append (ref)
m4 = re.findall (cregex4, term);
partofIDs = []
for ref in m4:
partofIDs.append (ref)
flatFilePrint (id, name, isaIDs, partofIDs)
else:
badElements += 1;
print >> sys.stderr, 'no match to m2...'
print >> sys.stderr, "---------------\n~np~%~/np~s\n------------------" ~np~%~/np~ term
print >> sys.stderr, 'goodElements ~np~%~/np~d' ~np~%~/np~ goodElements
print >> sys.stderr, 'badElements ~np~%~/np~d' ~np~%~/np~ badElements
#--------------------------------------
Script 1 - parseAssignmentsToFlatFileFromGoaProject.py
import sys
#-----------------------------------------------------------------------------------
def fixCanonicalName (rawName):
# for instance, trim 'YBR085W|ANC3' to 'YBR085W'
bar = rawName.find ('|')
if (bar < 0):
return rawName
return rawName [:bar]
#-----------------------------------------------------------------------------------
def fixGoID (rawID):
bar = rawID.find (':') + 1
return rawID [bar:]
#-----------------------------------------------------------------------------------
def readGoaXrefFile (filename):
lines = open (filename).read().split ('\n')
result = {}
for line in lines:
if (len (line) < 10):
continue
tokens = line.split ('\t')
ipi = tokens [2]
np = tokens [5]
semicolon = np.find (';')
if (semicolon >= 0):
np = np [:semicolon]
if (len (ipi) > 0 and len (np) > 0):
result [ipi] = np
return result
#-----------------------------------------------------------------------------------
if (len (sys.argv) != 3):
print 'error! parse <gene_associations file from GO> <goa xrefs file> '
sys.exit ()
associationFilename = sys.argv [1];
xrefsFilename = sys.argv [2]
species = 'Homo sapiens'
ipiToNPHash = readGoaXrefFile (xrefsFilename)
tester = 'IPI00099416'
print 'hash size: ~np~%~/np~d' ~np~%~/np~ len (ipiToNPHash)
print 'test map: ~np~%~/np~s -> NP_054861: ~np~%~/np~s ' ~np~%~/np~ (tester, ipiToNPHash [tester])
bioproc = open ('bioproc.txt', 'w')
molfunc = open ('molfunc.txt', 'w')
cellcomp = open ('cellcomp.txt', 'w')
bioproc.write ('(species=~np~%~/np~s) (type=Biological Process) (curator=GO)\n' ~np~%~/np~ species)
molfunc.write ('(species=~np~%~/np~s) (type=Molecular Function) (curator=GO)\n' ~np~%~/np~ species);
cellcomp.write ('(species=~np~%~/np~s) (type=Cellular Component) (curator=GO)\n' ~np~%~/np~ species);
lines=open(associationFilename).read().split('\n')
sys.stderr.write ('found ~np~%~/np~d lines\n' ~np~%~/np~ len (lines))
for line in lines:
if (line.find ('!') == 0 or len (line) < 2):
continue
tokens = line.split ('\t')
goOntology = tokens [8]
goIDraw = tokens [4]
goID = goIDraw.split (':')[1]
ipiName = fixCanonicalName (tokens [10])
if (len (ipiName) < 1):
continue
if (not ipiToNPHash.has_key (ipiName)):
continue
refseqName = ipiToNPHash [ipiName]
printName = refseqName
#printName = ipiName
if (ipiName == tester):
print '~np~%~/np~s (~np~%~/np~s) has go term ~np~%~/np~s' ~np~%~/np~ (tester, printName, goID)
if (goOntology == 'C'):
cellcomp.write ('~np~%~/np~s = ~np~%~/np~s\n' ~np~%~/np~ (printName, goID))
elif (goOntology == 'P'):
bioproc.write ('~np~%~/np~s = ~np~%~/np~s\n' ~np~%~/np~ (printName, goID))
elif (goOntology == 'F'):
molfunc.write ('~np~%~/np~s = ~np~%~/np~s\n' ~np~%~/np~ (printName, goID))
#-----------------------------------------------------------------------------------